

Prolonged unfolded protein reaction is involved in the induction of chronic myeloid leukemia cell death upon oprozomib treatment
- PMID: 33067904
- PMCID: PMC7780017
- DOI: 10.1111/cas.14696
Prolonged unfolded protein reaction is involved in the induction of chronic myeloid leukemia cell death upon oprozomib treatment
Abstract
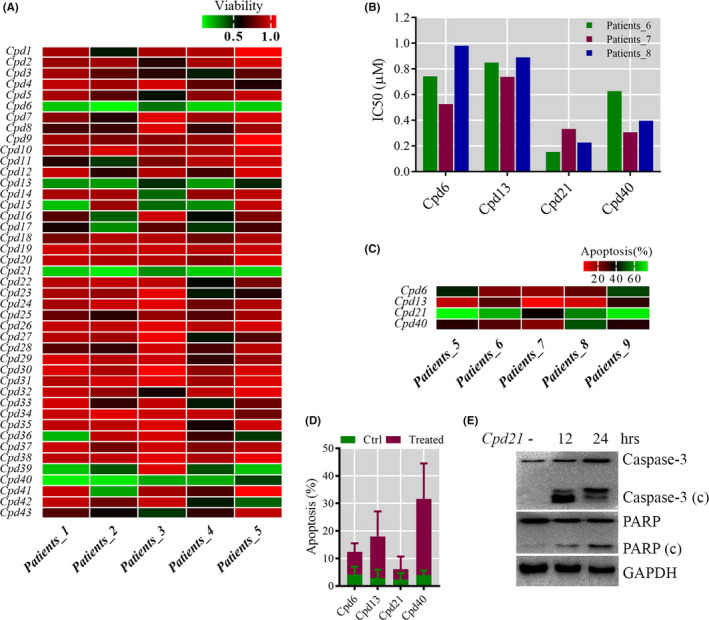
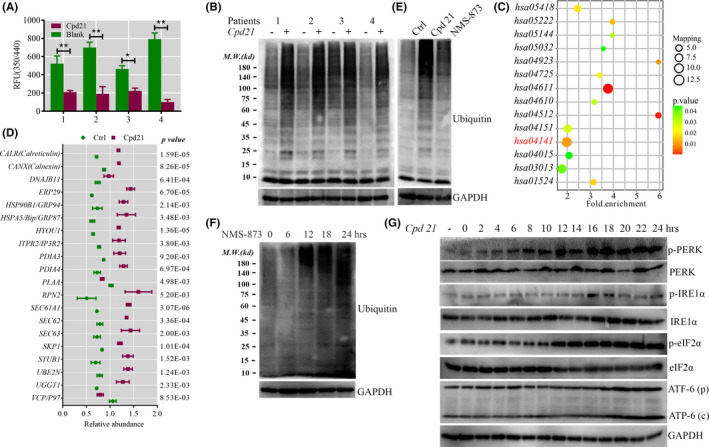
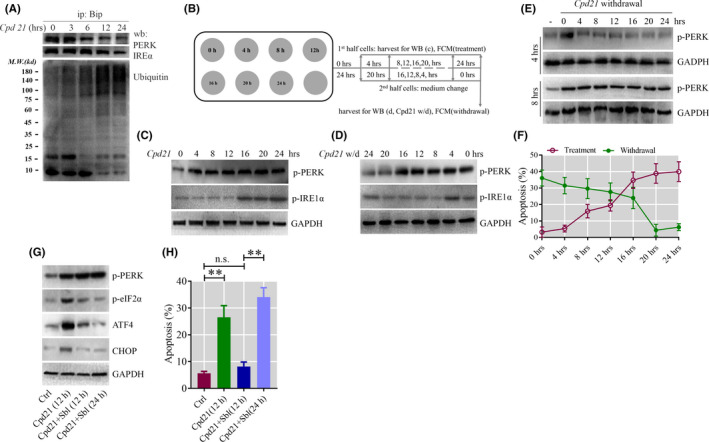
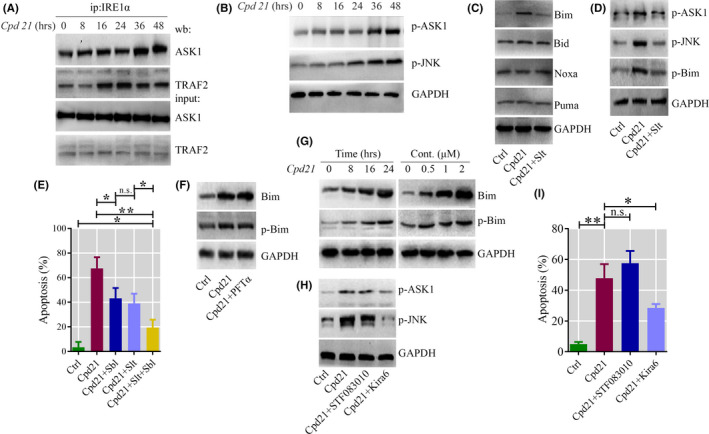
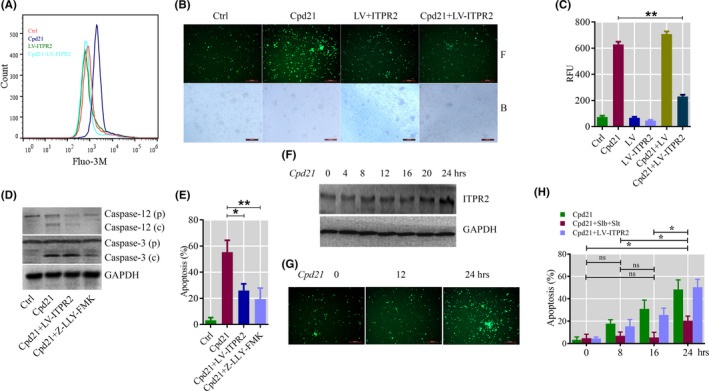
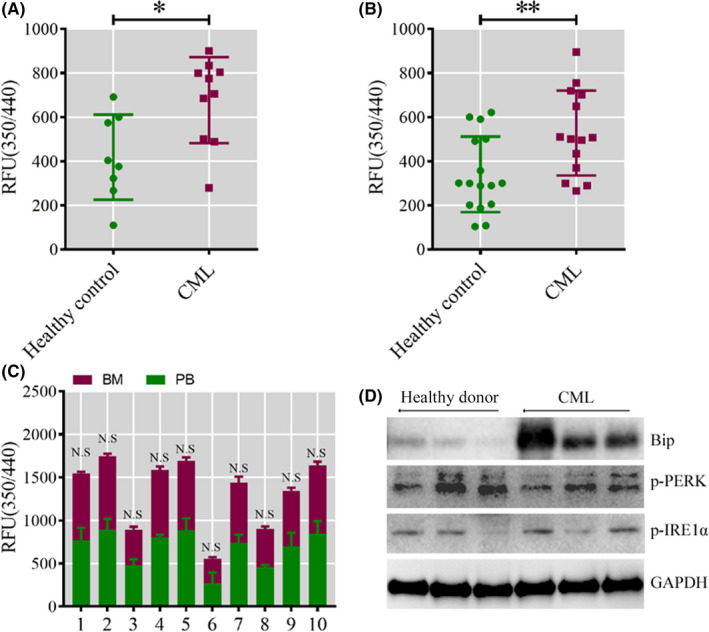
To select the most efficient chemical to induce apoptosis in leukemia cells, a multidrug screen was applied on bone marrow mononuclear cells from chronic myeloid leukemia (CML) patients. Oprozomib (Cpd 21) was chosen for the subsequent experiments. The isobaric tags for relative and absolute quantitation (iTRAQ) was then performed to identify the responsible pathway relative to apoptosis and the results showed that endoplasmic reticulum (ER) chaperones were upregulated. Apoptosis was attributed to a joint effect of calcium leakage andPERK and IRE1α phosphorylation. The PERK branch was responsible for the first wave of cell death that occurred within 24 hours. The later wave of apoptosis was mediated by IRE1α, which transmit apoptotic signals through the ASK-JNK-BIM axis. Release of Ca2+ from ER into cytosol resulted in activation of calpain, which, in turn, cleaved caspase-12. Our data also explained the selective killing effects of oprozomib on CML cells, which relied on proteasome activity. The present study demonstrated that prolonged inhibition of proteasome to trigger unfolded protein response could be an alternative strategy for treating CML in light of tyrosine kinase inhibitors resistance.
Keywords: CML; ER stress; apoptosis; proteasome inhibitor; unfolded protein reaction.
© 2020 The Authors. Cancer Science published by John Wiley & Sons Australia, Ltd on behalf of Japanese Cancer Association.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Chereda B, Melo JV. Natural course and biology of CML. Ann Hematol. 2015;94(Suppl. 2):S107‐S121. - PubMed
-
- Talati C, Pinilla‐Ibarz J. Resistance in chronic myeloid leukemia: definitions and novel therapeutic agents. Curr Opin Hematol. 2018;25:154‐161. - PubMed
-
- Ao N, Chen Q, Liu G. The small molecules targeting ubiquitin‐proteasome system for cancer therapy. Comb Chem High Throughput Screen. 2017;20:403‐413. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
