Local Sleep and Alzheimer's Disease Pathophysiology
- PMID: 33071726
- PMCID: PMC7538792
- DOI: 10.3389/fnins.2020.525970
Local Sleep and Alzheimer's Disease Pathophysiology
Abstract
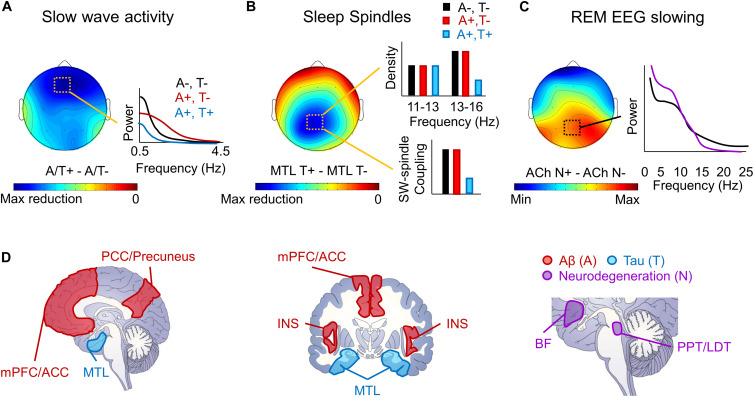
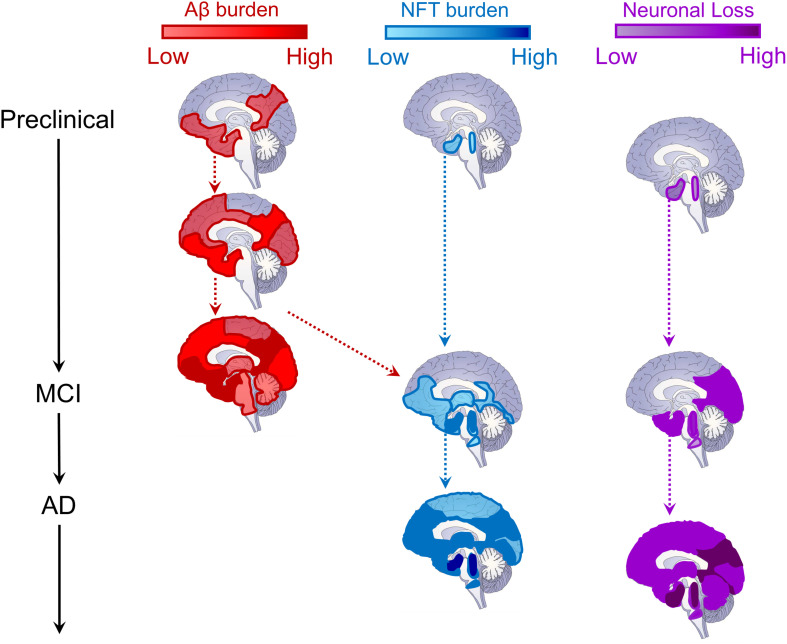
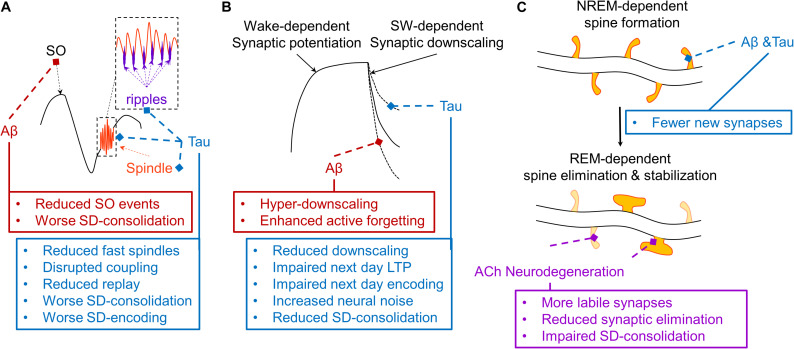
Even prior to the onset of the prodromal stages of Alzheimer's disease (AD), a constellation of sleep disturbances are apparent. A series of epidemiological studies indicate that multiple forms of these sleep disturbances are associated with increased risk for developing mild cognitive impairment (MCI) and AD, even triggering disease onset at an earlier age. Through the combination of causal manipulation studies in humans and rodents, as well as targeted examination of sleep disturbance with respect to AD biomarkers, mechanisms linking sleep disturbance to AD are beginning to emerge. In this review, we explore recent evidence linking local deficits in brain oscillatory function during sleep with local AD pathological burden and circuit-level dysfunction and degeneration. In short, three deficits in the local expression of sleep oscillations have been identified in relation to AD pathophysiology: (1) frequency-specific frontal deficits in slow wave expression during non-rapid eye movement (NREM) sleep, (2) deficits in parietal sleep spindle expression, and (3) deficits in the quality of electroencephalographic (EEG) desynchrony characteristic of REM sleep. These deficits are noteworthy since they differ from that seen in normal aging, indicating the potential presence of an abnormal aging process. How each of these are associated with β-amyloid (Aβ) and tau pathology, as well as neurodegeneration of circuits sensitive to AD pathophysiology, are examined in the present review, with a focus on the role of dysfunction within fronto-hippocampal and subcortical sleep-wake circuits. It is hypothesized that each of these local sleep deficits arise from distinct network-specific dysfunctions driven by regionally-specific accumulation of AD pathologies, as well as their associated neurodegeneration. Overall, the evolution of these local sleep deficits offer unique windows into the circuit-specific progression of distinct AD pathophysiological processes prior to AD onset, as well as their impact on brain function. This includes the potential erosion of sleep-dependent memory mechanisms, which may contribute to memory decline in AD. This review closes with a discussion of the remaining critical knowledge gaps and implications of this work for future mechanistic studies and studies implementing sleep-based treatment interventions.
Keywords: Alzheimer’s disease; REM sleep; beta-amyloid protein; neurodegeneration; sleep; sleep spindle; slow waves; tau and phospho-tau protein.
Copyright © 2020 Mander.
Figures