

Near-Single-Cell Proteomics Profiling of the Proximal Tubular and Glomerulus of the Normal Human Kidney
- PMID: 33072769
- PMCID: PMC7533534
- DOI: 10.3389/fmed.2020.00499
Near-Single-Cell Proteomics Profiling of the Proximal Tubular and Glomerulus of the Normal Human Kidney
Erratum in
-
Corrigendum: Near-Single-Cell Proteomics Profiling of the Proximal Tubular and Glomerulus of the Normal Human Kidney.Front Med (Lausanne). 2021 Jan 12;7:625788. doi: 10.3389/fmed.2020.625788. eCollection 2020. Front Med (Lausanne). 2021. PMID: 33511148 Free PMC article.
Abstract
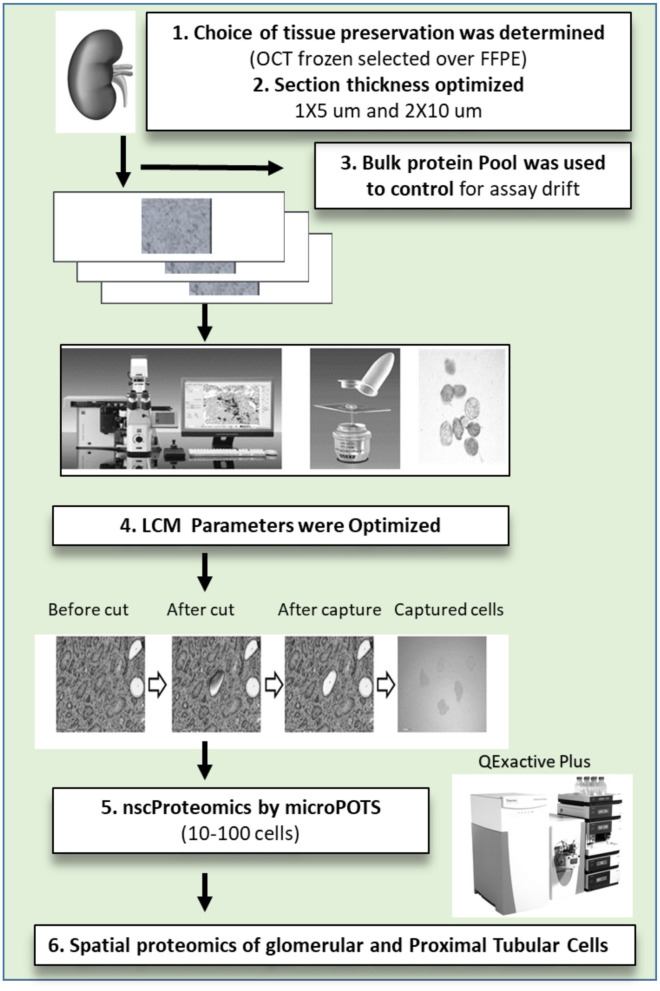
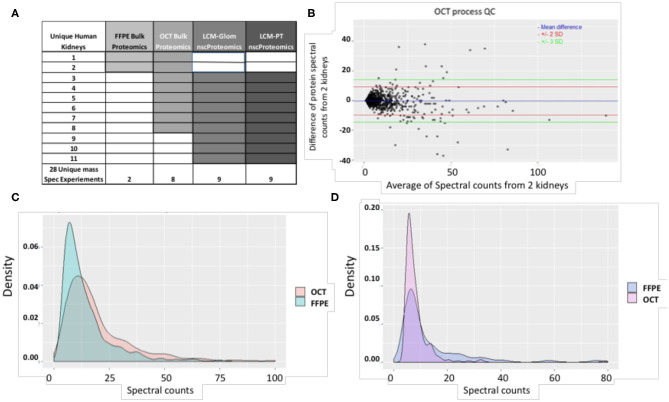
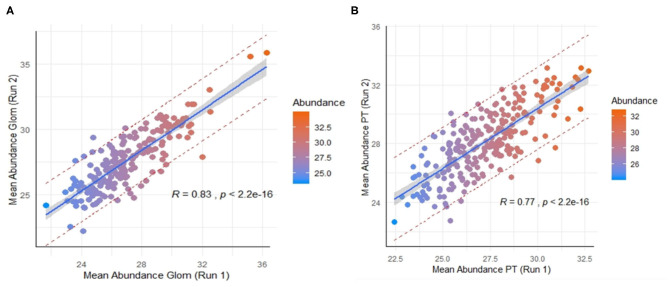
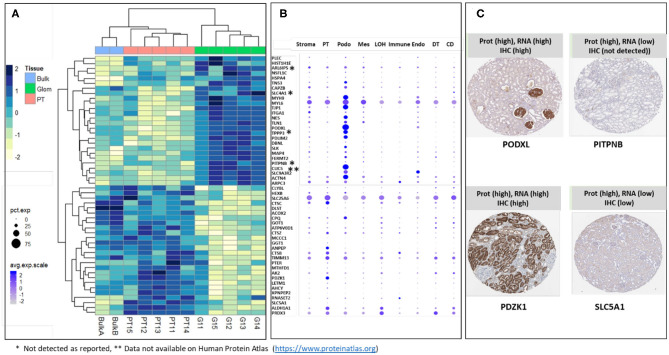
Molecular assessments at the single cell level can accelerate biological research by providing detailed assessments of cellular organization and tissue heterogeneity in both disease and health. The human kidney has complex multi-cellular states with varying functionality, much of which can now be completely harnessed with recent technological advances in tissue proteomics at a near single-cell level. We discuss the foundational steps in the first application of this mass spectrometry (MS) based proteomics method for analysis of sub-sections of the normal human kidney, as part of the Kidney Precision Medicine Project (KPMP). Using ~30-40 laser captured micro-dissected kidney cells, we identified more than 2,500 human proteins, with specificity to the proximal tubular (PT; n = 25 proteins) and glomerular (Glom; n = 67 proteins) regions of the kidney and their unique metabolic functions. This pilot study provides the roadmap for application of our near-single-cell proteomics workflow for analysis of other renal micro-compartments, on a larger scale, to unravel perturbations of renal sub-cellular function in the normal kidney as well as different etiologies of acute and chronic kidney disease.
Keywords: glomerulus; kidney; mass spectrometry; proteomics; single cell analysis.
Copyright © 2020 Sigdel, Piehowski, Roy, Liberto, Hansen, Swensen, Zhao, Zhu, Rashmi, Schroeder, Damm, Sur, Luo, Yang, Qian, Sarwal and the Kidney Precision Medicine Project (KPMP) Consortium.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases