

Sickle Cell Disease-Genetics, Pathophysiology, Clinical Presentation and Treatment
- PMID: 33072979
- PMCID: PMC7510211
- DOI: 10.3390/ijns5020020
Sickle Cell Disease-Genetics, Pathophysiology, Clinical Presentation and Treatment
Abstract
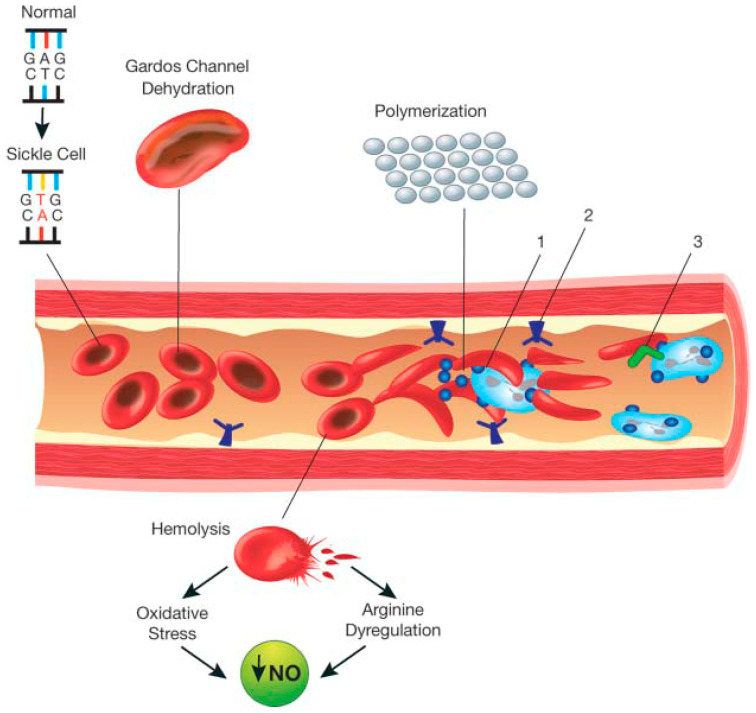
Sickle cell disease (SCD) is a monogenetic disorder due to a single base-pair point mutation in the β-globin gene resulting in the substitution of the amino acid valine for glutamic acid in the β-globin chain. Phenotypic variation in the clinical presentation and disease outcome is a characteristic feature of the disorder. Understanding the pathogenesis and pathophysiology of the disorder is central to the choice of therapeutic development and intervention. In this special edition for newborn screening for haemoglobin disorders, it is pertinent to describe the genetic, pathologic and clinical presentation of sickle cell disease as a prelude to the justification for screening. Through a systematic review of the literature using search terms relating to SCD up till 2019, we identified relevant descriptive publications for inclusion. The scope of this review is mainly an overview of the clinical features of pain, the cardinal symptom in SCD, which present following the drop in foetal haemoglobin as young as five to six months after birth. The relative impact of haemolysis and small-vessel occlusive pathology remains controversial, a combination of features probably contribute to the different pathologies. We also provide an overview of emerging therapies in SCD.
Keywords: acute chest syndrome; anaemia; bone marrow transplant; end-organ damage; foetal haemoglobin; gene therapy for haemoglobinopathies; haemolysis; hydroxyurea/hydroxycarbamide; pathophysiology; sickle cell disease (SCD); vaso-occlusive crisis.
© 2019 by the authors.
Conflict of interest statement
Conflicts of InterestB.P.D.I. receives educational grant from Global Therapeutics, Pfizer, Novartis plc Cyclerion and honorarium from Novartis plc. L.L.H. is a consultant for Hilton Publishing, Pfizer, AstraZeneca, Emmaus, Emmi Solutions, and University of Cincinnati.
Figures
References
-
- Ballas S.K., Kesen M.R., Goldberg M.F., Lutty G.A., Dampier C., Osunkwo I., Wang W.C., Hoppe C., Hagar W., Darbari D.S., et al. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Management. Sci. World J. 2012;2012:949535. doi: 10.1100/2012/949535. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
