

The First Year Experience of Newborn Screening for Pompe Disease in California
- PMID: 33073007
- PMCID: PMC7422988
- DOI: 10.3390/ijns6010009
The First Year Experience of Newborn Screening for Pompe Disease in California
Abstract
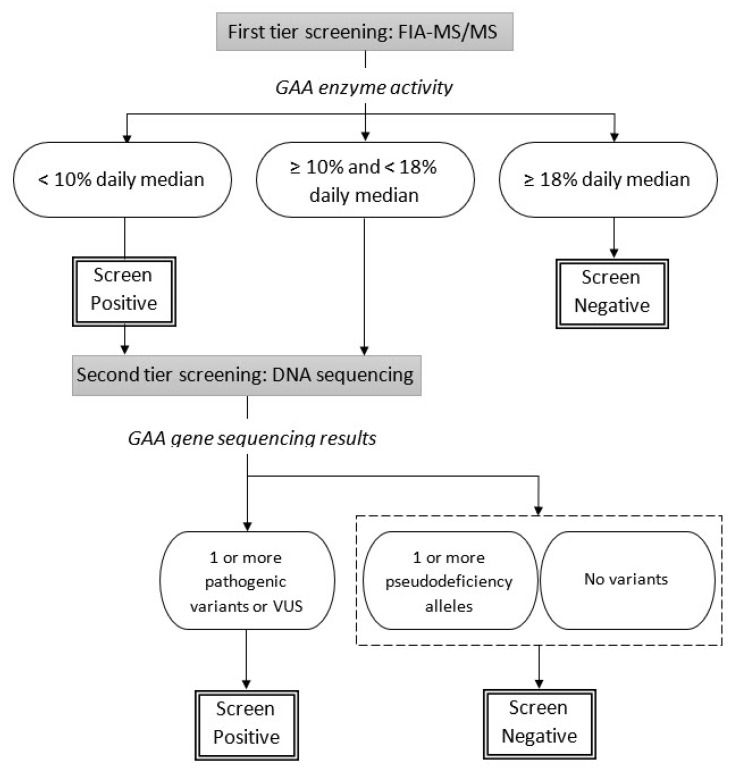
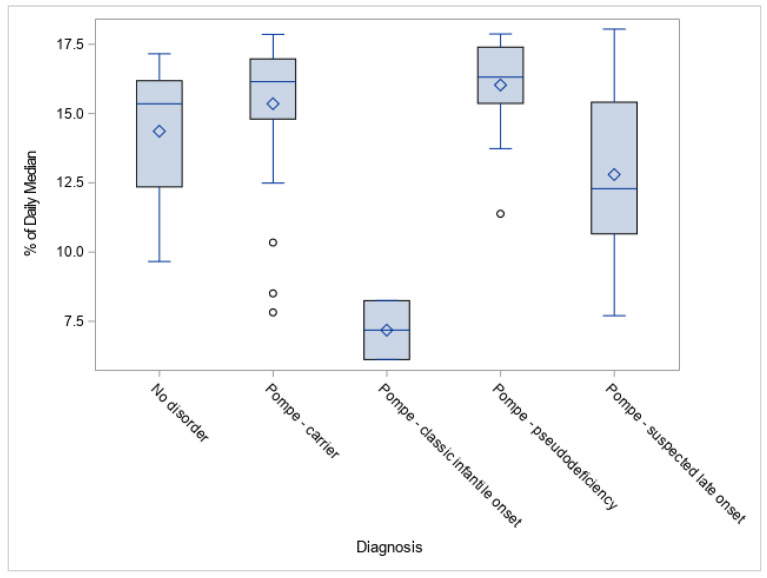
The California Department of Public Health started universal newborn screening for Pompe disease in August 2018 with a two-tier process including: (1) acid alpha-glucosidase (GAA) enzyme activity assay followed by, (2) GAA gene sequencing analysis. This study examines results from the first year of screening in a large and diverse screening population. With 453,152 screened newborns, the birth prevalence and GAA enzyme activity associated with various types of Pompe disease classifications are described. The frequency of GAA gene mutations and allele variants are reported. Of 88 screen positives, 18 newborns were resolved as Pompe disease, including 2 classic infantile-onset and 16 suspected late-onset form. The c.-32-13T>G variant was the most common pathogenic mutation reported. African American and Asian/Pacific Islander newborns had higher allele frequencies for both pathogenic and pseudodeficiency variants. After the first year of Pompe disease screening in California, the disease distribution in the population is now better understood. With the ongoing long-term follow-up system currently in place, our understanding of the complex genotype-phenotype relationships will become more evident in the future, and this should help us better understand the clinical significance of identified cases.
Keywords: California; Pompe disease; newborn screening.
© 2020 by the authors.
Conflict of interest statement
Conflicts of InterestThe authors declare no conflict of interest.
Figures
References
-
- Reuser A.J.J., Hirschhorn R., Kroos M.A. Pompe Disease: Glycogen storage disease type II: Acid α-glucosidase (acid maltase) deficiency. In: Valle D., Antonarakis S., Ballabio A., editors. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York, NY, USA: 2018.
-
- Martiniuk F., Chen A., Mack A., Arvanitopoulos E., Chen Y., Rom W.N., Codd W.J., Hanna B., Alcabes P., Raben N., et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am. J. Med. Genet. 1998;79:69–72. doi: 10.1002/(SICI)1096-8628(19980827)79:1<69::AID-AJMG16>3.0.CO;2-K. - DOI - PubMed
-
- Bashan N., Potashnik R., Barash V., Gutman A., Moses S.W. Glycogen storage disease type II in Israel. Isr. J. Med. Sci. 1988;24:224–227. - PubMed
-
- Scott C.R., Elliott S., Buroker N., Thomas L.I., Keutzer J., Glass M., Gelb M.H., Turecek F. Identification of infants at risk for developing Fabry, Pompe, or mucopolysaccharidosis-I from newborn blood spots by tandem mass spectrometry. J. Pediatr. 2013;163:498–503. doi: 10.1016/j.jpeds.2013.01.031. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
