

Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches
- PMID: 33076254
- PMCID: PMC7587536
- DOI: 10.3390/molecules25204723
Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches
Abstract
Virtual screening (VS) is an outstanding cornerstone in the drug discovery pipeline. A variety of computational approaches, which are generally classified as ligand-based (LB) and structure-based (SB) techniques, exploit key structural and physicochemical properties of ligands and targets to enable the screening of virtual libraries in the search of active compounds. Though LB and SB methods have found widespread application in the discovery of novel drug-like candidates, their complementary natures have stimulated continued efforts toward the development of hybrid strategies that combine LB and SB techniques, integrating them in a holistic computational framework that exploits the available information of both ligand and target to enhance the success of drug discovery projects. In this review, we analyze the main strategies and concepts that have emerged in the last years for defining hybrid LB + SB computational schemes in VS studies. Particularly, attention is focused on the combination of molecular similarity and docking, illustrating them with selected applications taken from the literature.
Keywords: combined strategies; drug discovery; ligand-based techniques; structure-based methods; virtual screening.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
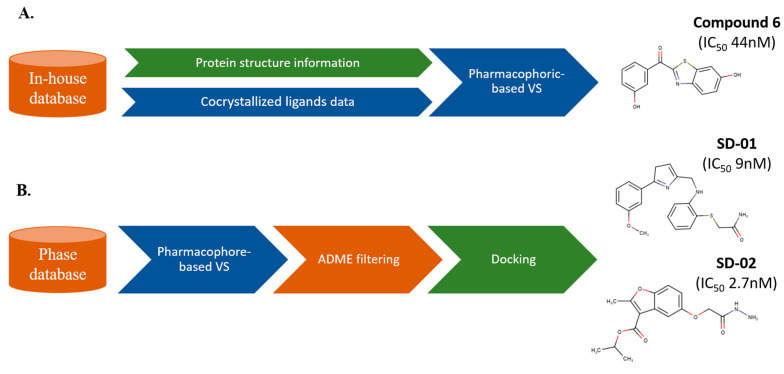
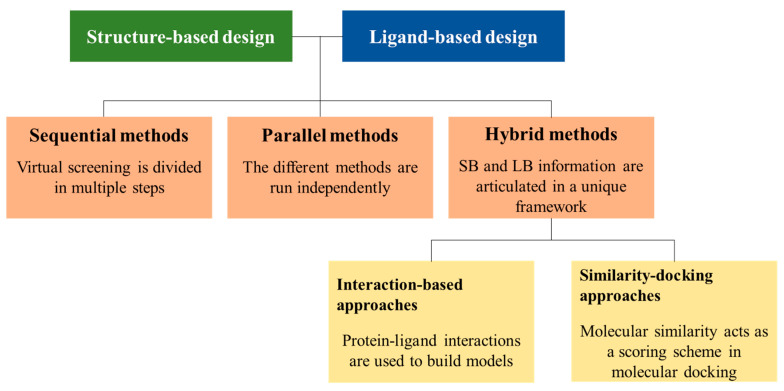
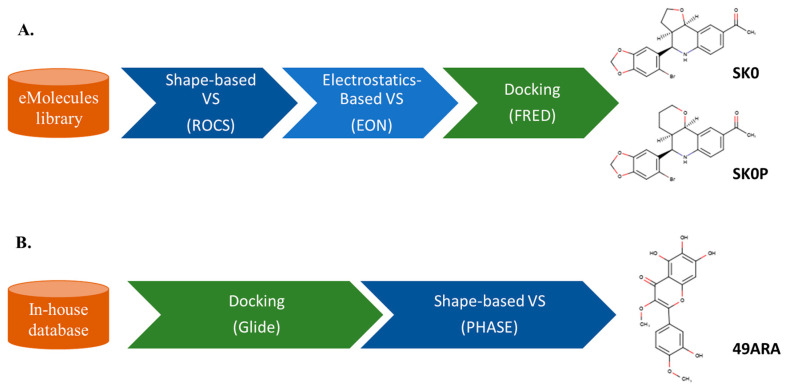
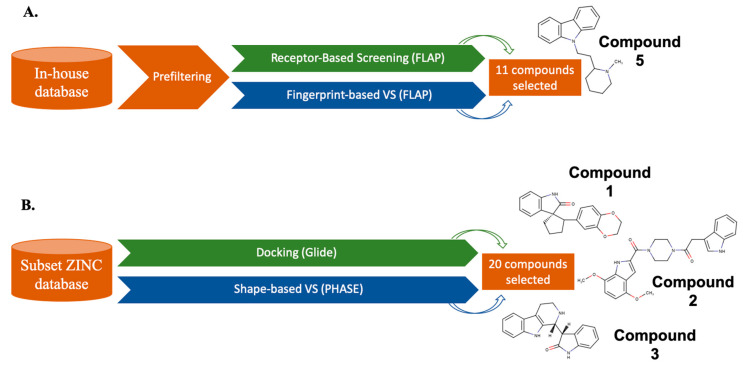
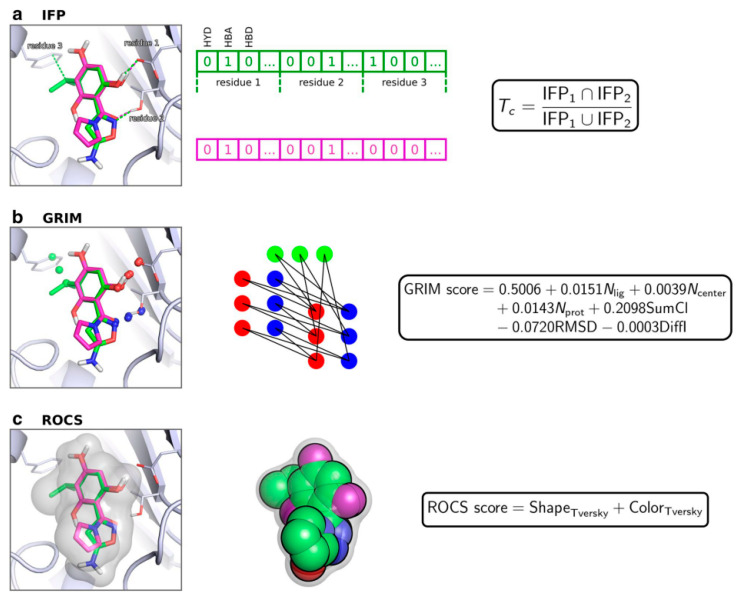
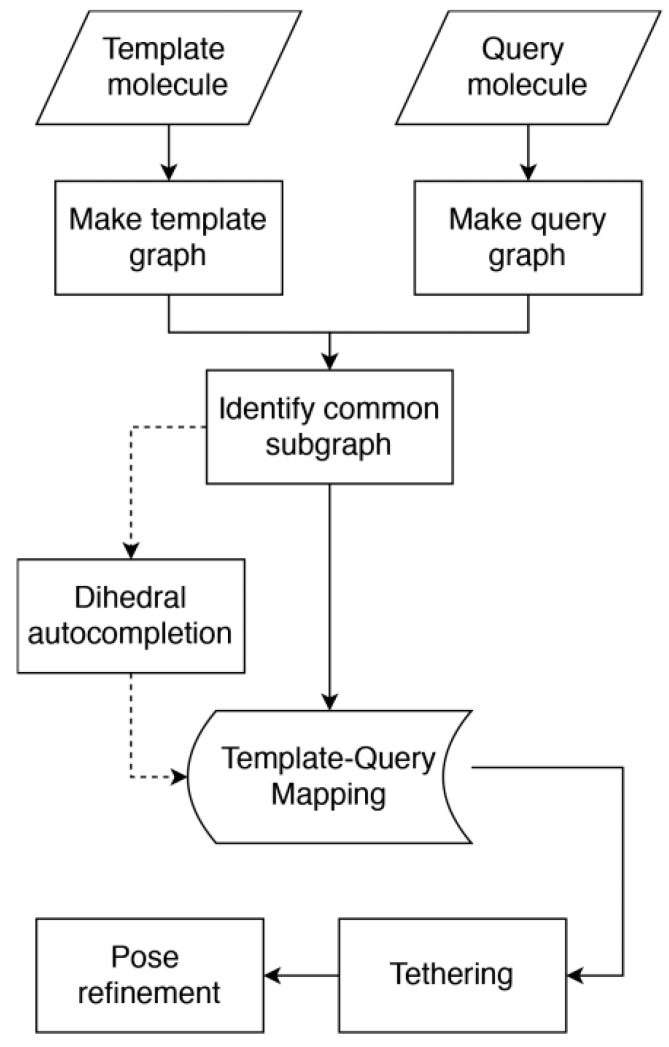
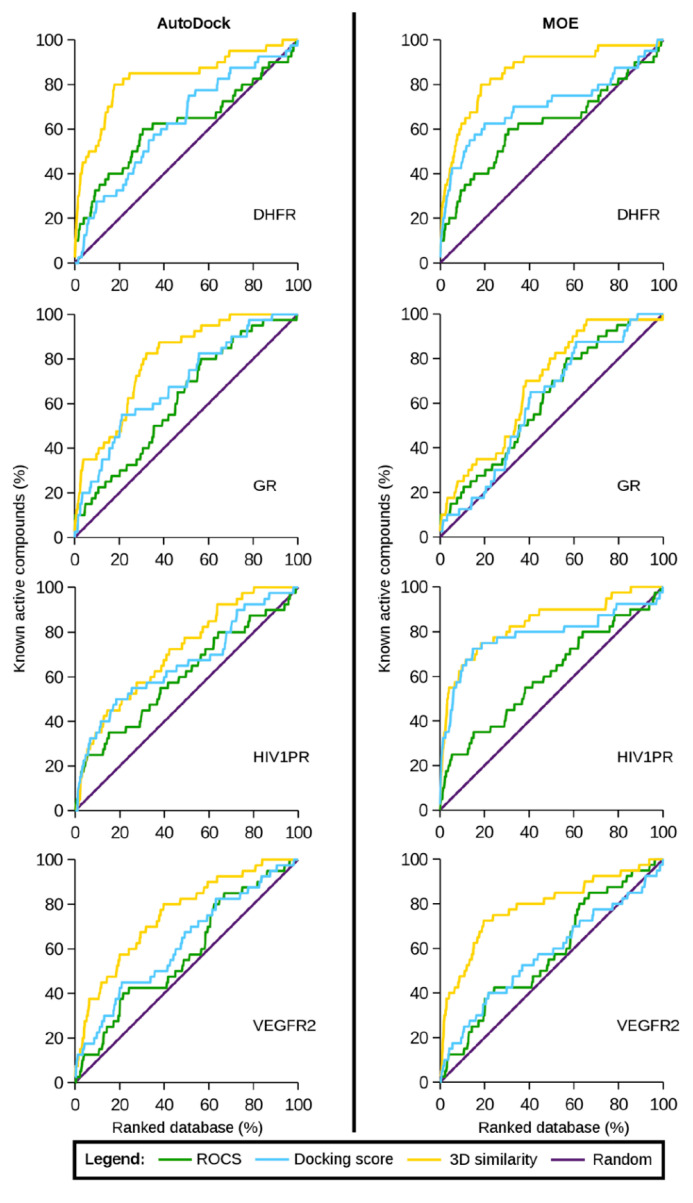
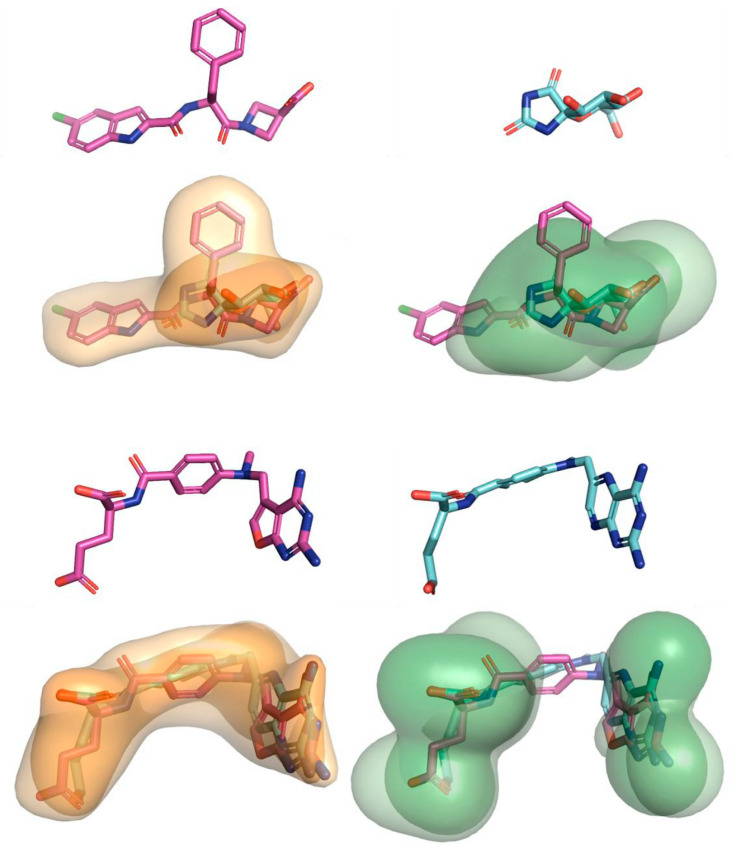
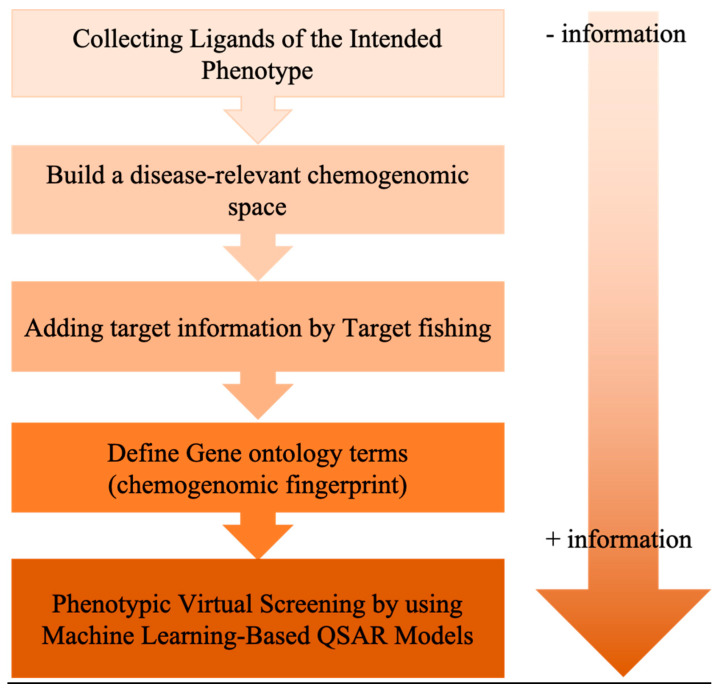
References
-
- Free Energy Calculations . In: Theory and Applications in Chemistry and Biology. Chipot C., Pohorille A., editors. Springer; Berlin/Heidelberg, Germany: 2007.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
