

A Novel CCT5 Missense Variant Associated with Early Onset Motor Neuropathy
- PMID: 33076433
- PMCID: PMC7589105
- DOI: 10.3390/ijms21207631
A Novel CCT5 Missense Variant Associated with Early Onset Motor Neuropathy
Abstract
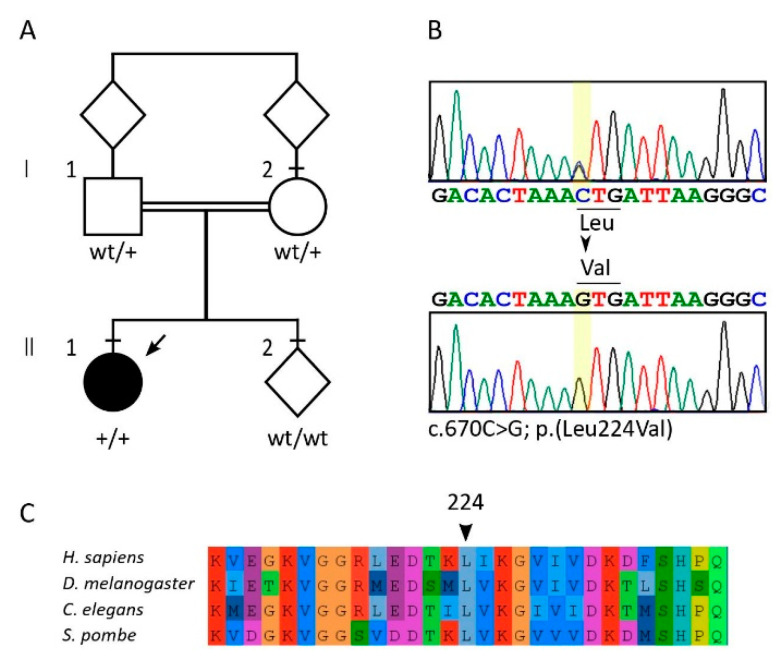
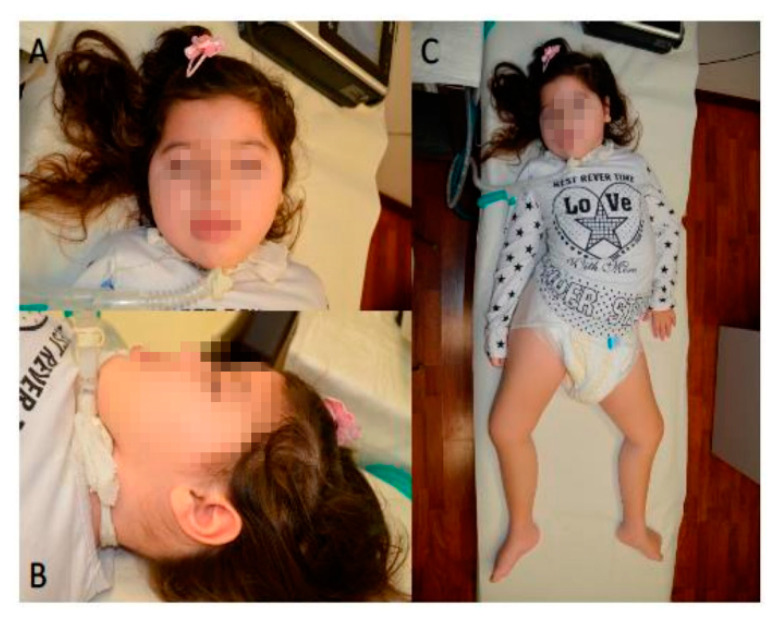
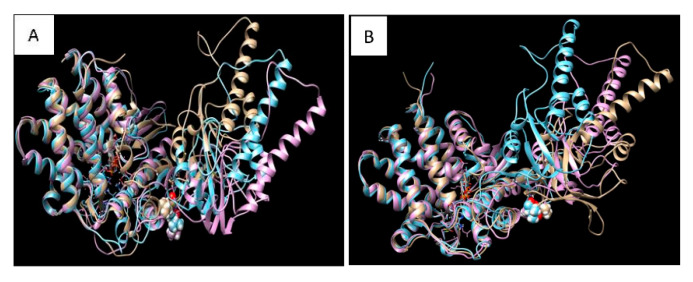
Diseases associated with acquired or genetic defects in members of the chaperoning system (CS) are increasingly found and have been collectively termed chaperonopathies. Illustrative instances of genetic chaperonopathies involve the genes for chaperonins of Groups I (e.g., Heat shock protein 60, Hsp60) and II (e.g., Chaperonin Containing T-Complex polypeptide 1, CCT). Examples of the former are hypomyelinating leukodystrophy 4 (HLD4 or MitCHAP60) and hereditary spastic paraplegia (SPG13). A distal sensory mutilating neuropathy has been linked to a mutation [p.(His147Arg)] in subunit 5 of the CCT5 gene. Here, we describe a new possibly pathogenic variant [p.(Leu224Val)] of the same subunit but with a different phenotype. This yet undescribed disease affects a girl with early onset demyelinating neuropathy and a severe motor disability. By whole exome sequencing (WES), we identified a homozygous CCT5 c.670C>G p.(Leu224Val) variant in the CCT5 gene. In silico 3D-structure analysis and bioinformatics indicated that this variant could undergo abnormal conformation and could be pathogenic. We compared the patient's clinical, neurophysiological and laboratory data with those from patients carrying p.(His147Arg) in the equatorial domain. Our patient presented signs and symptoms absent in the p.(His147Arg) cases. Molecular dynamics simulation and modelling showed that the Leu224Val mutation that occurs in the CCT5 intermediate domain near the apical domain induces a conformational change in the latter. Noteworthy is the striking difference between the phenotypes putatively linked to mutations in the same CCT subunit but located in different structural domains, offering a unique opportunity for elucidating their distinctive roles in health and disease.
Keywords: CCT5; chaperoning system; chaperonins; genetic chaperonopathies; genetic variants; motor neuropathy; mutation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Macario A.J.L., Conway de Macario E., Cappello F. The Chaperonopathies: Diseases with Defective Molecular Chaperones. Springer; Dordrecht, The Netherlands: Heidelberg, Germany: New York, NY, USA: London, UK: 2013.
-
- Bouhouche A., Benomar A., Bouslam N., Chkili T., Yahyaoui M. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 2006;43:441–443. doi: 10.1136/jmg.2005.039230. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
