

Exploration of the sputum methylome and omics deconvolution by quadratic programming in molecular profiling of asthma and COPD: the road to sputum omics 2.0
- PMID: 33076907
- PMCID: PMC7574293
- DOI: 10.1186/s12931-020-01544-4
Exploration of the sputum methylome and omics deconvolution by quadratic programming in molecular profiling of asthma and COPD: the road to sputum omics 2.0
Abstract
Background: To date, most studies involving high-throughput analyses of sputum in asthma and COPD have focused on identifying transcriptomic signatures of disease. No whole-genome methylation analysis of sputum cells has been performed yet. In this context, the highly variable cellular composition of sputum has potential to confound the molecular analyses.
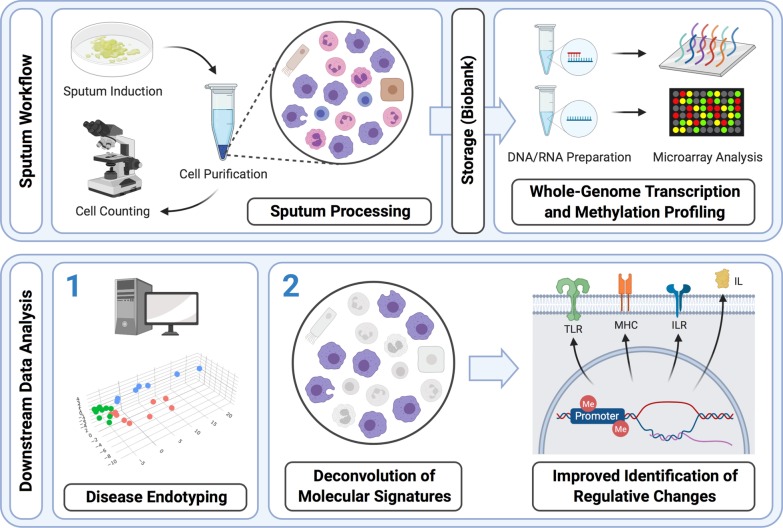
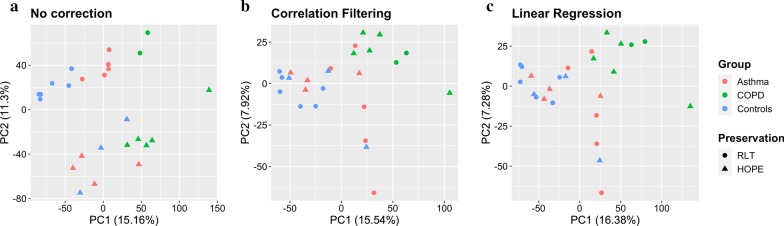
Methods: Whole-genome transcription (Agilent Human 4 × 44 k array) and methylation (Illumina 450 k BeadChip) analyses were performed on sputum samples of 9 asthmatics, 10 healthy and 10 COPD subjects. RNA integrity was checked by capillary electrophoresis and used to correct in silico for bias conferred by RNA degradation during biobank sample storage. Estimates of cell type-specific molecular profiles were derived via regression by quadratic programming based on sputum differential cell counts. All analyses were conducted using the open-source R/Bioconductor software framework.
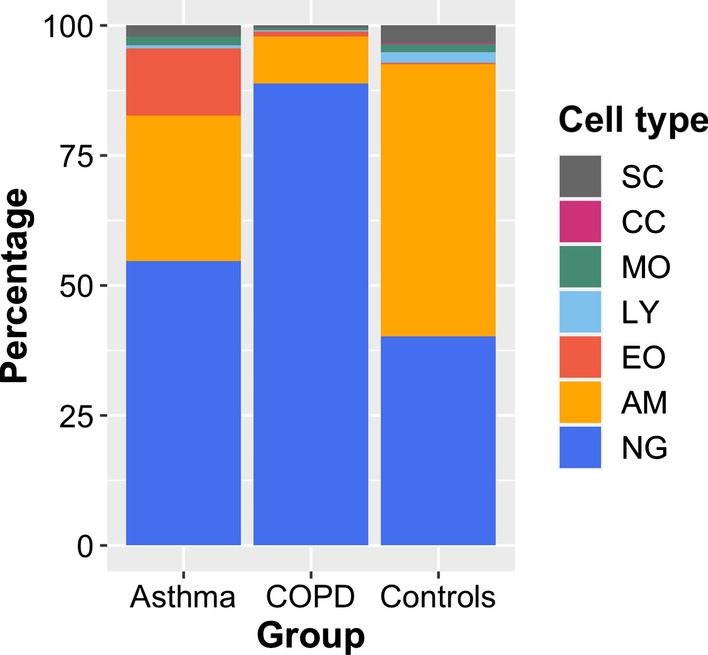
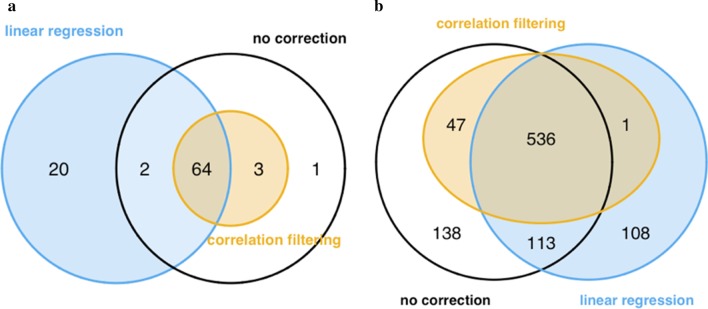
Results: A linear regression step was found to perform well in removing RNA degradation-related bias among the main principal components of the gene expression data, increasing the number of genes detectable as differentially expressed in asthma and COPD sputa (compared to controls). We observed a strong influence of the cellular composition on the results of mixed-cell sputum analyses. Exemplarily, upregulated genes derived from mixed-cell data in asthma were dominated by genes predominantly expressed in eosinophils after deconvolution. The deconvolution, however, allowed to perform differential expression and methylation analyses on the level of individual cell types and, though we only analyzed a limited number of biological replicates, was found to provide good estimates compared to previously published data about gene expression in lung eosinophils in asthma. Analysis of the sputum methylome indicated presence of differential methylation in genomic regions of interest, e.g. mapping to a number of human leukocyte antigen (HLA) genes related to both major histocompatibility complex (MHC) class I and II molecules in asthma and COPD macrophages. Furthermore, we found the SMAD3 (SMAD family member 3) gene, among others, to lie within differentially methylated regions which has been previously reported in the context of asthma.
Conclusions: In this methodology-oriented study, we show that methylation profiling can be easily integrated into sputum analysis workflows and exhibits a strong potential to contribute to the profiling and understanding of pulmonary inflammation. Wherever RNA degradation is of concern, in silico correction can be effective in improving both sensitivity and specificity of downstream analyses. We suggest that deconvolution methods should be integrated in sputum omics analysis workflows whenever possible in order to facilitate the unbiased discovery and interpretation of molecular patterns of inflammation.
Keywords: Asthma; Biobanking; COPD; Deconvolution; Degradation; Methylome; Omics; RNA; Sputum; Transcriptome.
Conflict of interest statement
The authors declare they have no competing interests pertaining this work.
Figures

References
-
- Kuo CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, Sousa A, Corfield J, Djukanovic R, Lutter R, et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J. 2017;49:1602135. doi: 10.1183/13993003.02135-2016. - DOI - PubMed
-
- Govoni M, Bassi M, Vezzoli S, Lucci G, Emirova A, Nandeuil MA, Petruzzelli S, Jellema GL, Afolabi EK, Colgan B, et al. Sputum and blood transcriptomics characterisation of the inhaled PDE4 inhibitor CHF6001 on top of triple therapy in patients with chronic bronchitis. Respir Res. 2020;21:72. doi: 10.1186/s12931-020-1329-y. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
