

Implications of metabolism-driven myeloid dysfunctions in cancer therapy
- PMID: 33077904
- PMCID: PMC7570408
- DOI: 10.1038/s41423-020-00556-w
Implications of metabolism-driven myeloid dysfunctions in cancer therapy
Abstract
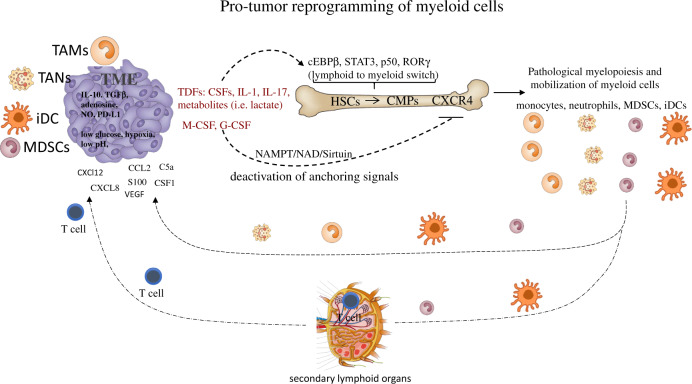
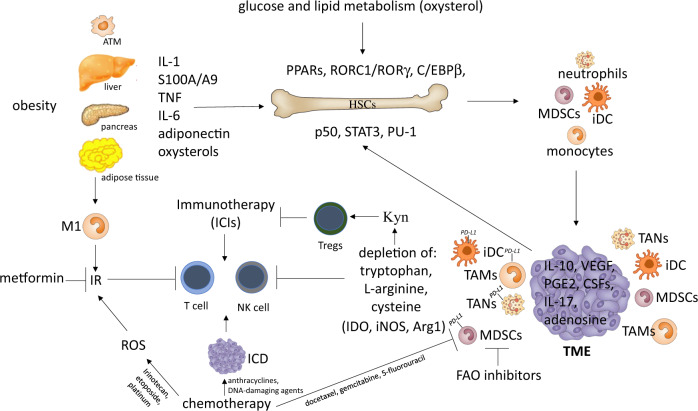
Immune homeostasis is maintained by an adequate balance of myeloid and lymphoid responses. In chronic inflammatory states, including cancer, this balance is lost due to dramatic expansion of myeloid progenitors that fail to mature to functional inflammatory neutrophils, macrophages, and dendritic cells (DCs), thus giving rise to a decline in the antitumor effector lymphoid response. Cancer-related inflammation orchestrates the production of hematopoietic growth factors and cytokines that perpetuate recruitment and activation of myeloid precursors, resulting in unresolved and chronic inflammation. This pathologic inflammation creates profound alterations in the intrinsic cellular metabolism of the myeloid progenitor pool, which is amplified by competition for essential nutrients and by hypoxia-induced metabolic rewiring at the tumor site. Therefore, persistent myelopoiesis and metabolic dysfunctions contribute to the development of cancer, as well as to the severity of a broad range of diseases, including metabolic syndrome and autoimmune and infectious diseases. The aims of this review are to (1) define the metabolic networks implicated in aberrant myelopoiesis observed in cancer patients, (2) discuss the mechanisms underlying these clinical manifestations and the impact of metabolic perturbations on clinical outcomes, and (3) explore new biomarkers and therapeutic strategies to restore immunometabolism and differentiation of myeloid cells towards an effector phenotype to increase host antitumor immunity. We propose that the profound metabolic alterations and associated transcriptional changes triggered by chronic and overactivated immune responses in myeloid cells represent critical factors influencing the balance between therapeutic efficacy and immune-related adverse effects (irAEs) for current therapeutic strategies, including immune checkpoint inhibitor (ICI) therapy.
Keywords: Cancer therapy; Metabolism; Myeloid-derived suppressor cells; Myelopoiesis; Tumor-associated macrophages.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy.Cancer Immunol Immunother. 2015 Jan;64(1):1-13. doi: 10.1007/s00262-014-1639-3. Epub 2014 Nov 29. Cancer Immunol Immunother. 2015. PMID: 25432147 Free PMC article. Review.
-
Myelopoiesis during Solid Cancers and Strategies for Immunotherapy.Cells. 2021 Apr 21;10(5):968. doi: 10.3390/cells10050968. Cells. 2021. PMID: 33919157 Free PMC article. Review.
-
Promotion of Expansion and Differentiation of Hematopoietic Stem Cells by Interleukin-27 into Myeloid Progenitors to Control Infection in Emergency Myelopoiesis.PLoS Pathog. 2016 Mar 18;12(3):e1005507. doi: 10.1371/journal.ppat.1005507. eCollection 2016 Mar. PLoS Pathog. 2016. PMID: 26991425 Free PMC article.
-
Transcriptional regulation of myeloid-derived suppressor cells.J Leukoc Biol. 2015 Dec;98(6):913-22. doi: 10.1189/jlb.4RI0515-204R. Epub 2015 Sep 3. J Leukoc Biol. 2015. PMID: 26337512 Free PMC article. Review.
-
Specific and Complex Reprogramming of Cellular Metabolism in Myeloid Cells during Innate Immune Responses.Cell Metab. 2017 Jul 5;26(1):142-156. doi: 10.1016/j.cmet.2017.06.001. Cell Metab. 2017. PMID: 28683282 Review.
Cited by
-
Injectable cellular vesicle-based bone meal for inflammatory bone defect repair through restoring immune homeostasis.Theranostics. 2025 Mar 18;15(10):4465-4480. doi: 10.7150/thno.110795. eCollection 2025. Theranostics. 2025. PMID: 40225579 Free PMC article.
-
Evolution and Targeting of Myeloid Suppressor Cells in Cancer: A Translational Perspective.Cancers (Basel). 2022 Jan 20;14(3):510. doi: 10.3390/cancers14030510. Cancers (Basel). 2022. PMID: 35158779 Free PMC article. Review.
-
c-Rel-dependent monocytes are potent immune suppressor cells in cancer.J Leukoc Biol. 2022 Oct;112(4):845-859. doi: 10.1002/JLB.1MA0422-518RR. Epub 2022 Jun 13. J Leukoc Biol. 2022. PMID: 35694784 Free PMC article.
-
RORγ Bridges Cancer-Driven Lipid Dysmetabolism and Myeloid Immunosuppression.Cancer Discov. 2025 Jul 3;15(7):1505-1525. doi: 10.1158/2159-8290.CD-24-0199. Cancer Discov. 2025. PMID: 40066819 Free PMC article.
-
The systemic-level repercussions of cancer-associated inflammation mediators produced in the tumor microenvironment.Front Endocrinol (Lausanne). 2022 Aug 22;13:929572. doi: 10.3389/fendo.2022.929572. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36072935 Free PMC article. Review.
References
-
- Escamilla-Tilch M, et al. The interplay between pathogen-associated and danger-associated molecular patterns: an inflammatory code in cancer? Immunol. Cell Biol. 2013;91:601–610. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
