

Exome sequencing implicates genetic disruption of prenatal neuro-gliogenesis in sporadic congenital hydrocephalus
- PMID: 33077954
- PMCID: PMC7871900
- DOI: 10.1038/s41591-020-1090-2
Exome sequencing implicates genetic disruption of prenatal neuro-gliogenesis in sporadic congenital hydrocephalus
Abstract
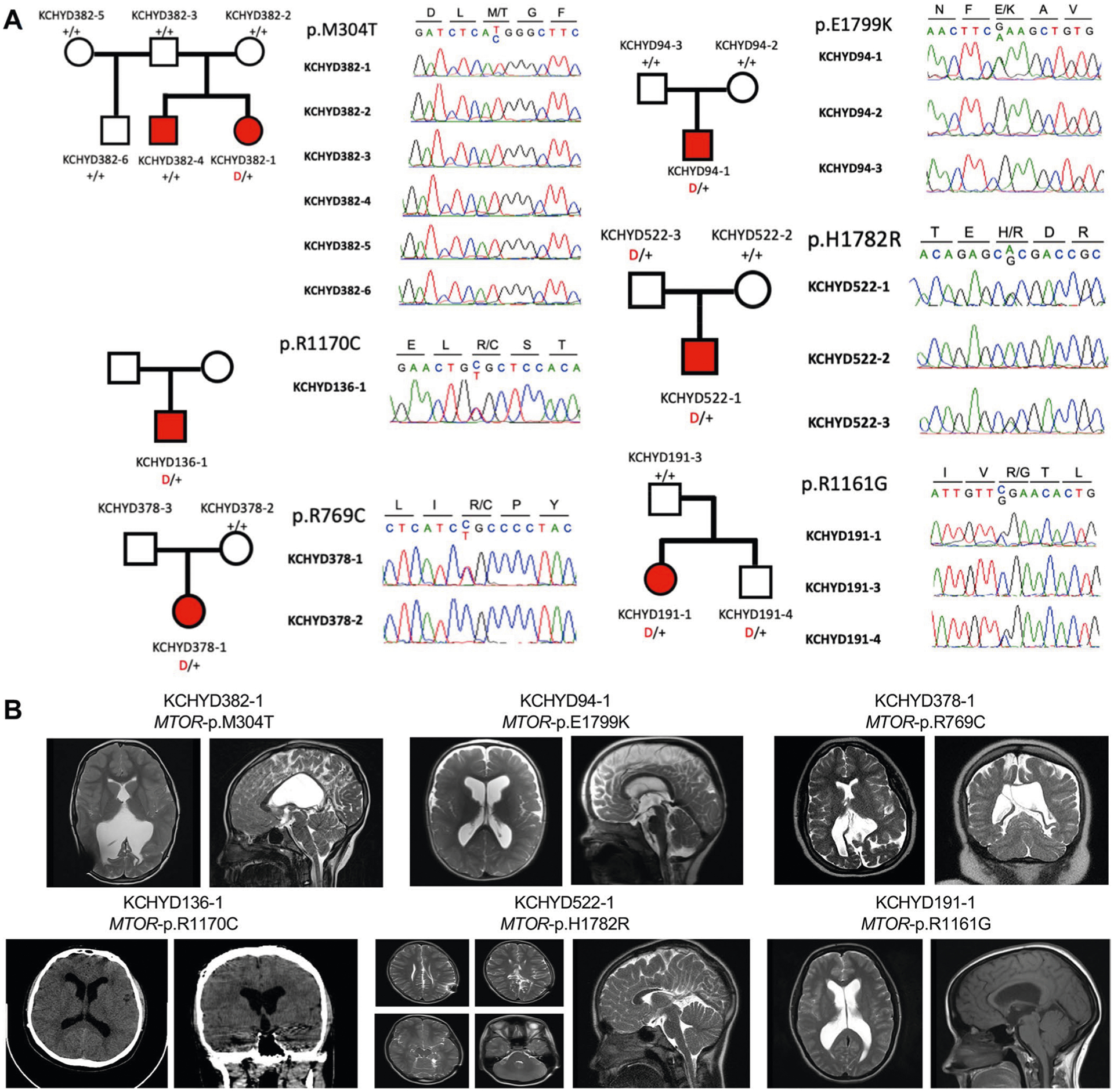
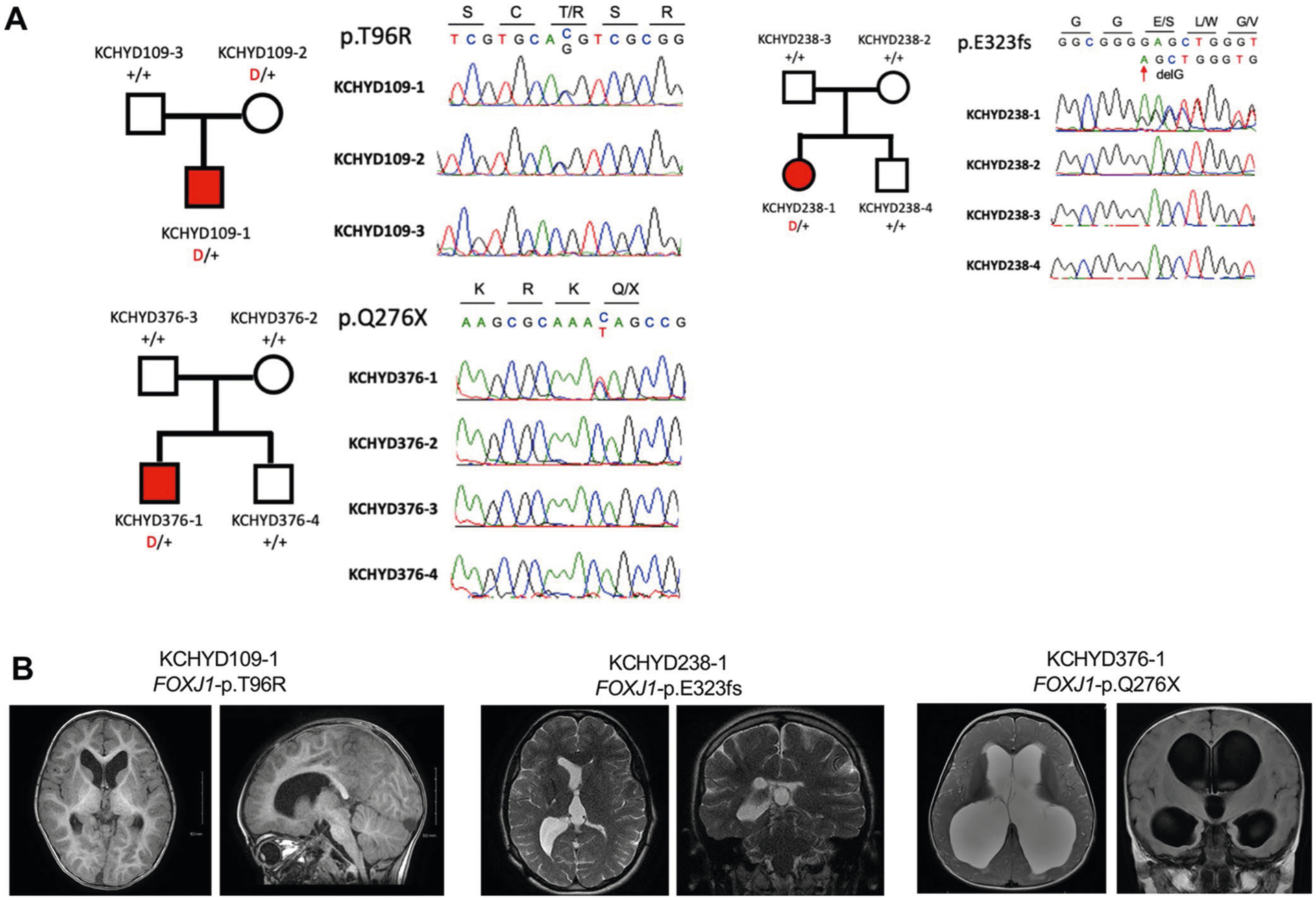
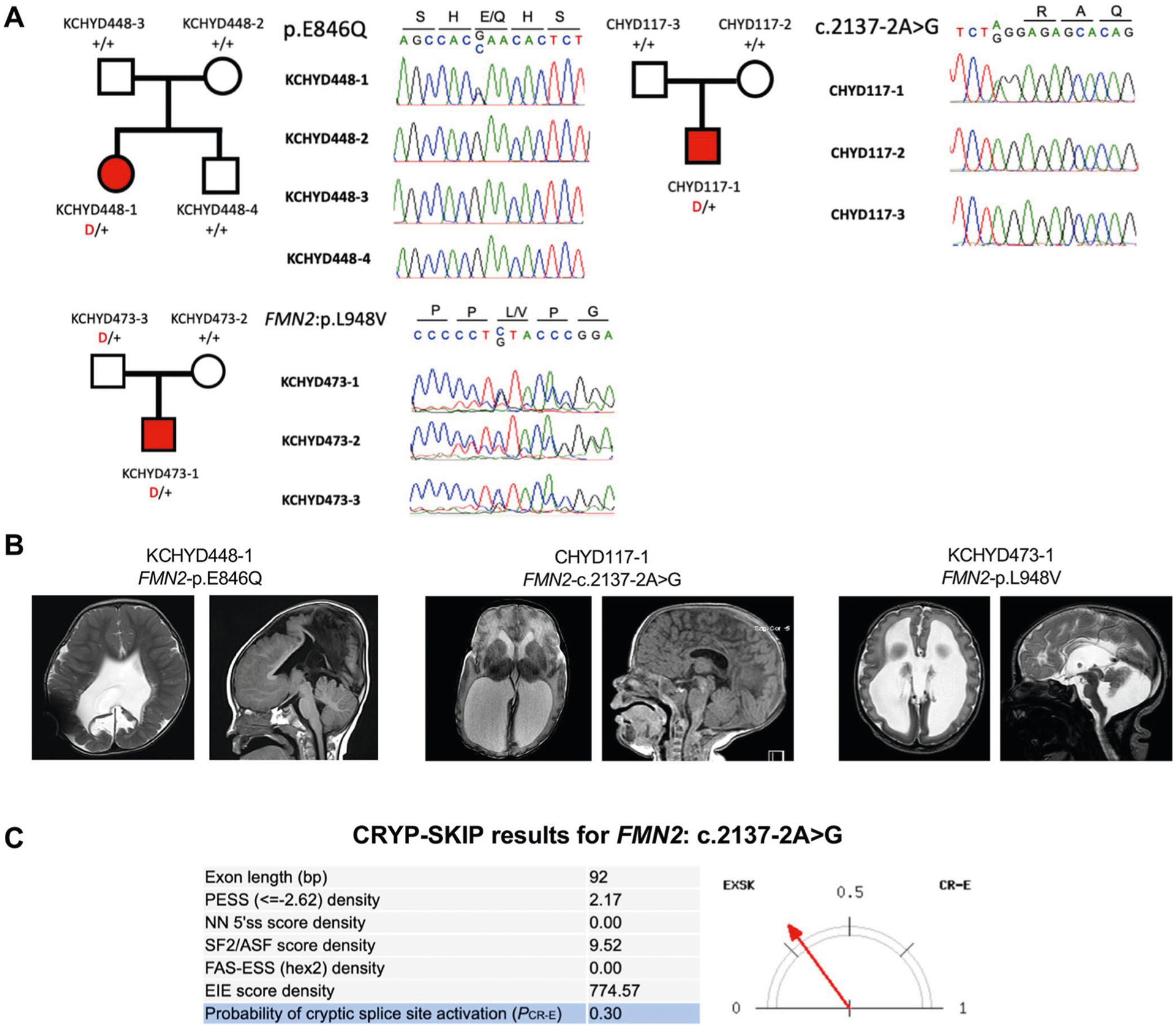
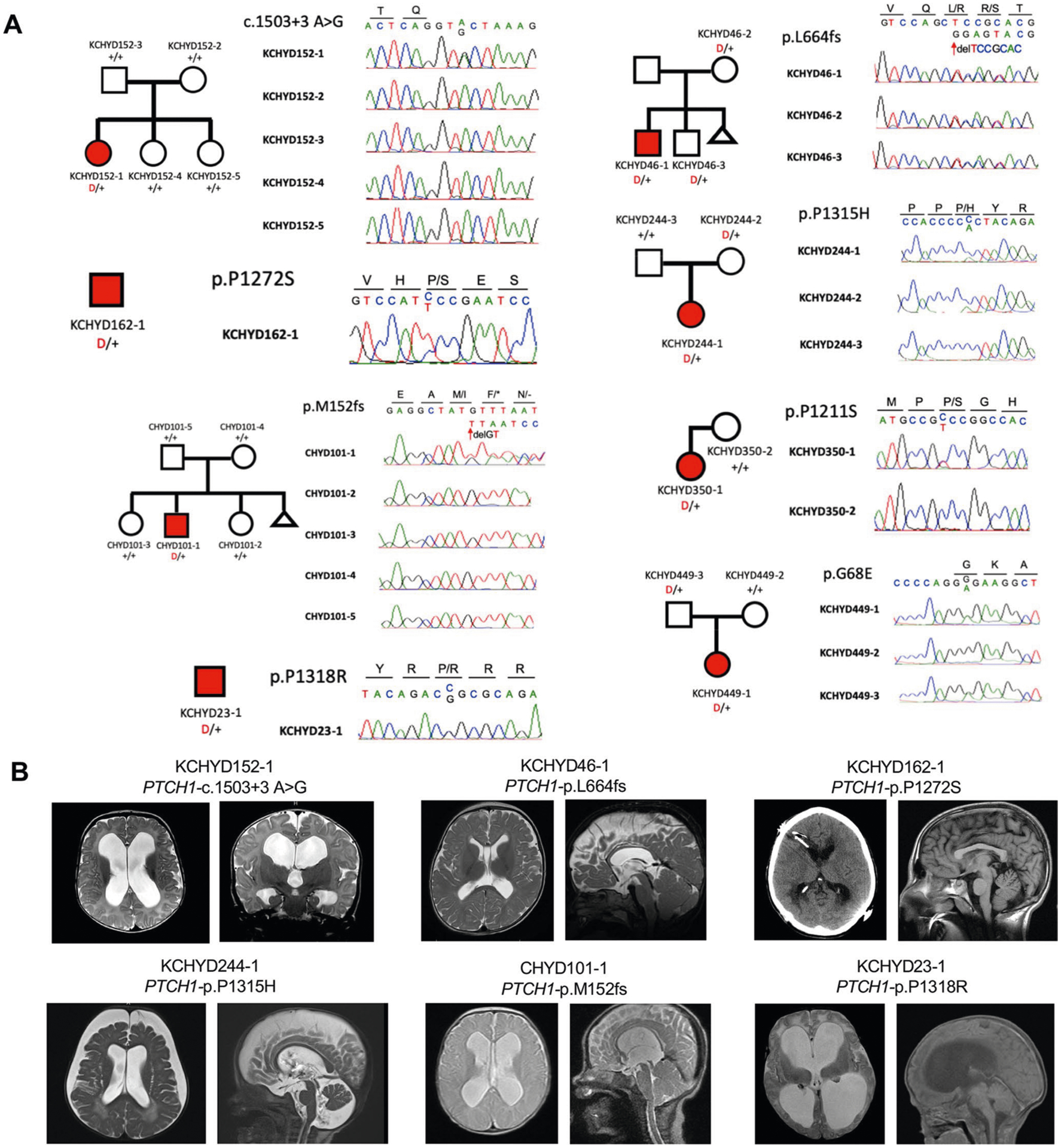
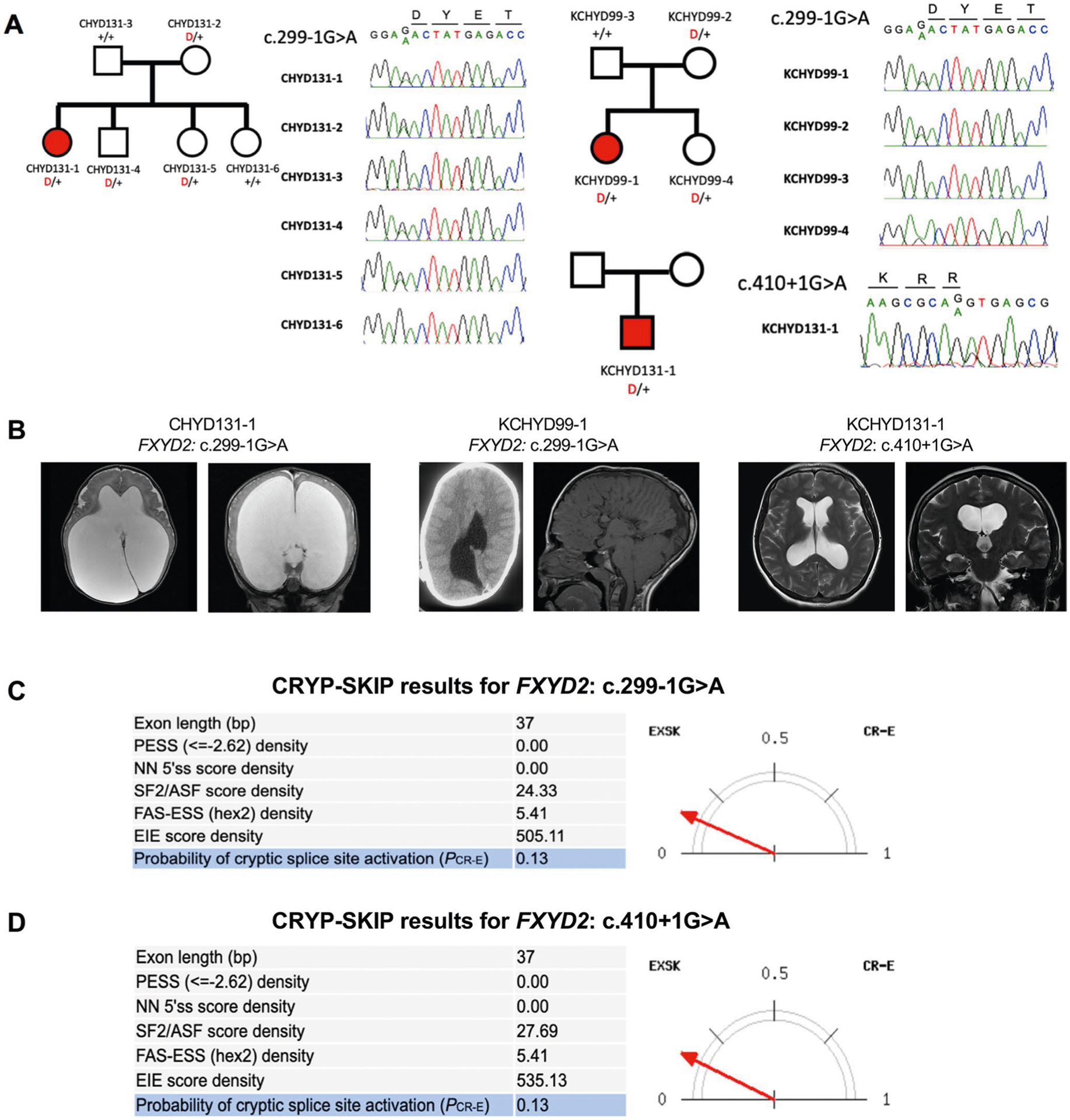
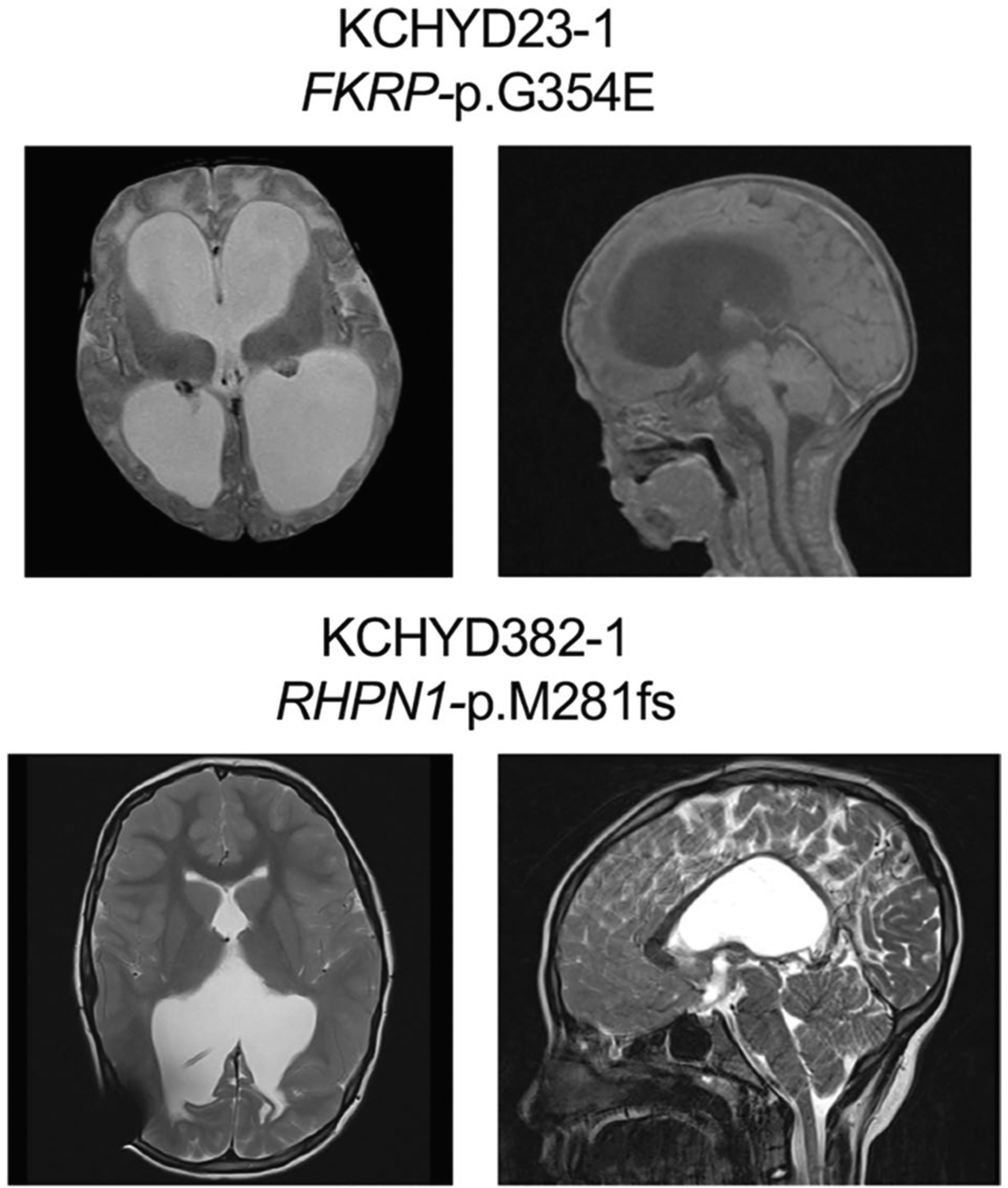
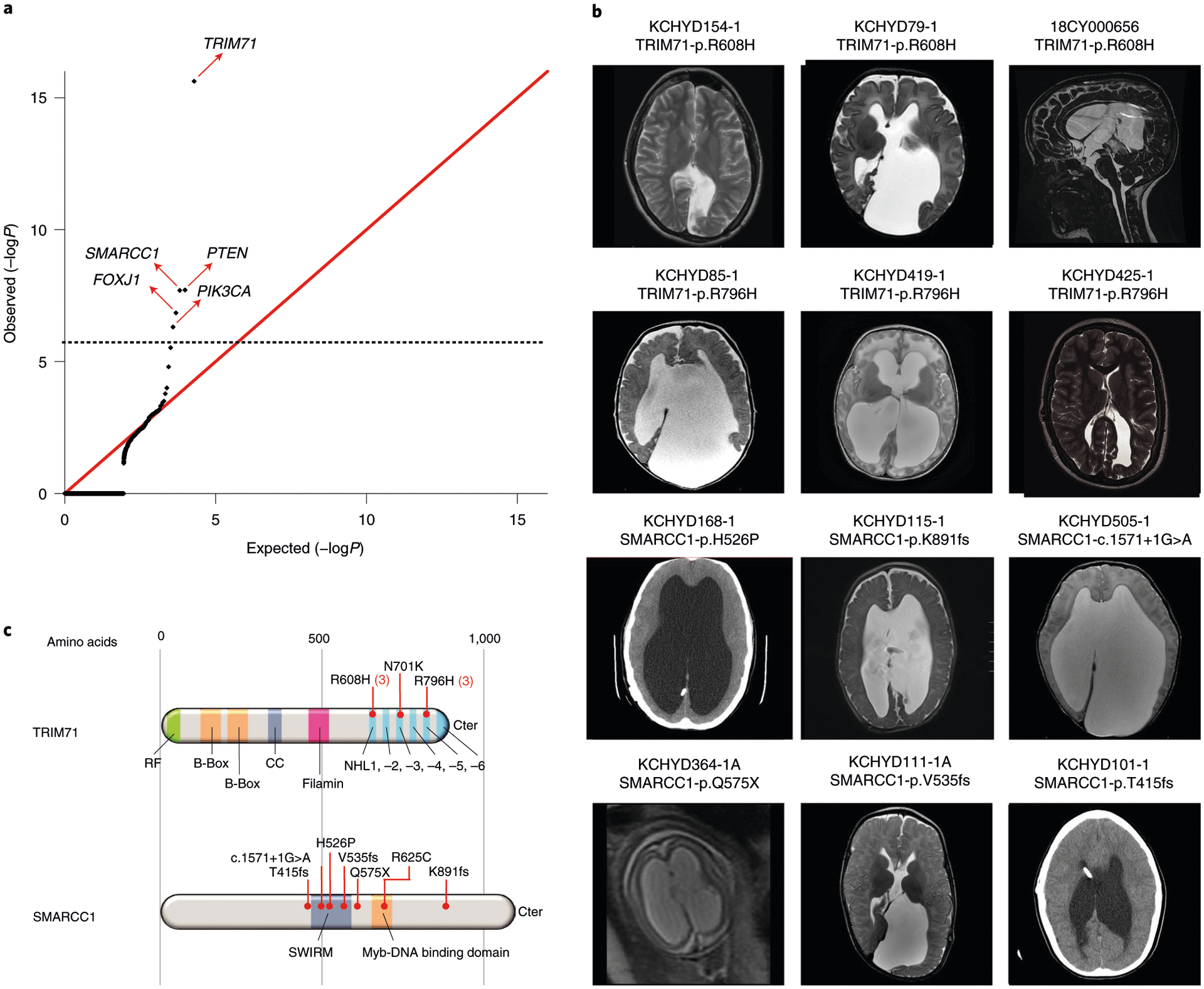
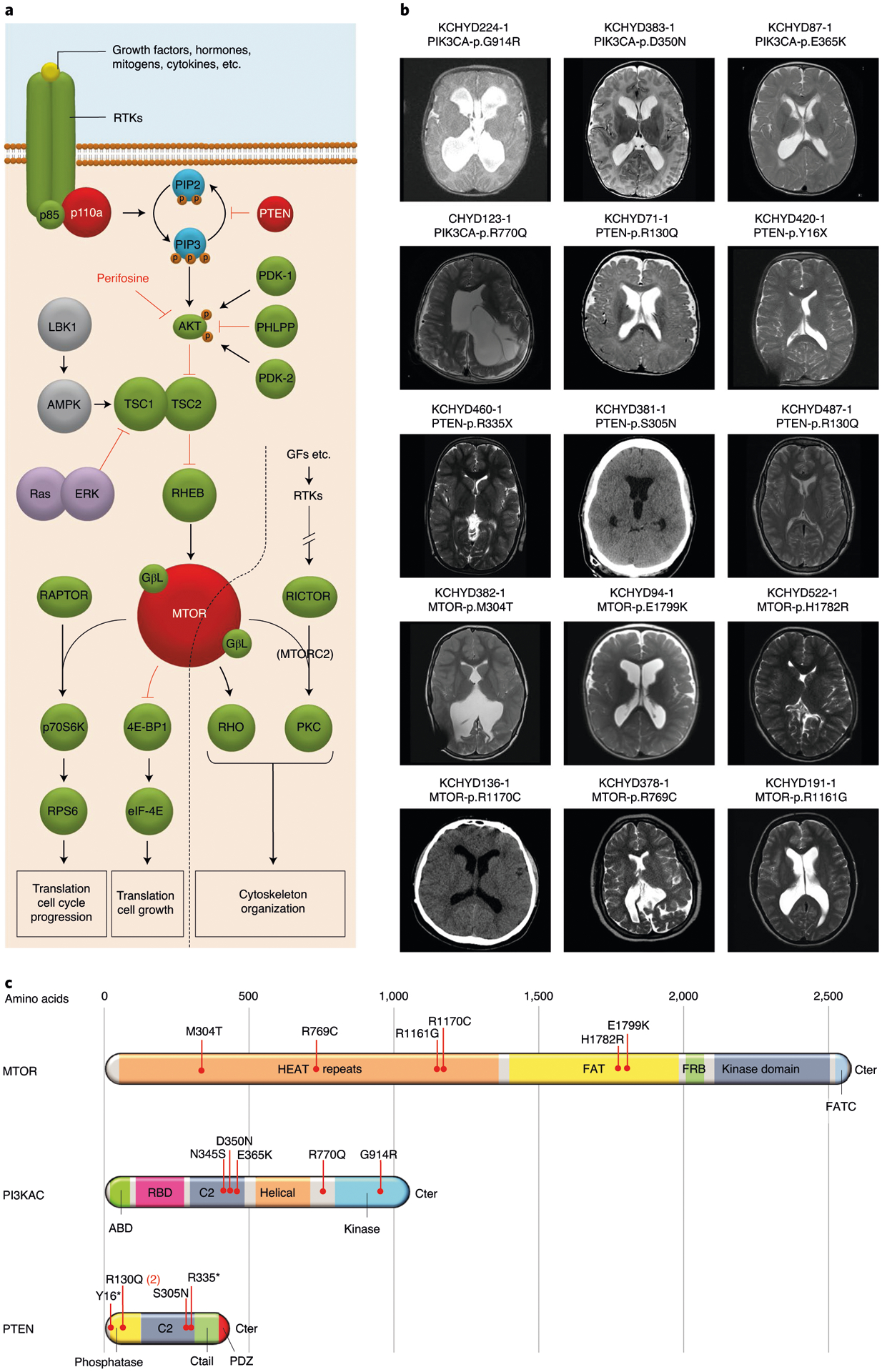
Congenital hydrocephalus (CH), characterized by enlarged brain ventricles, is considered a disease of excessive cerebrospinal fluid (CSF) accumulation and thereby treated with neurosurgical CSF diversion with high morbidity and failure rates. The poor neurodevelopmental outcomes and persistence of ventriculomegaly in some post-surgical patients highlight our limited knowledge of disease mechanisms. Through whole-exome sequencing of 381 patients (232 trios) with sporadic, neurosurgically treated CH, we found that damaging de novo mutations account for >17% of cases, with five different genes exhibiting a significant de novo mutation burden. In all, rare, damaging mutations with large effect contributed to ~22% of sporadic CH cases. Multiple CH genes are key regulators of neural stem cell biology and converge in human transcriptional networks and cell types pertinent for fetal neuro-gliogenesis. These data implicate genetic disruption of early brain development, not impaired CSF dynamics, as the primary pathomechanism of a significant number of patients with sporadic CH.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
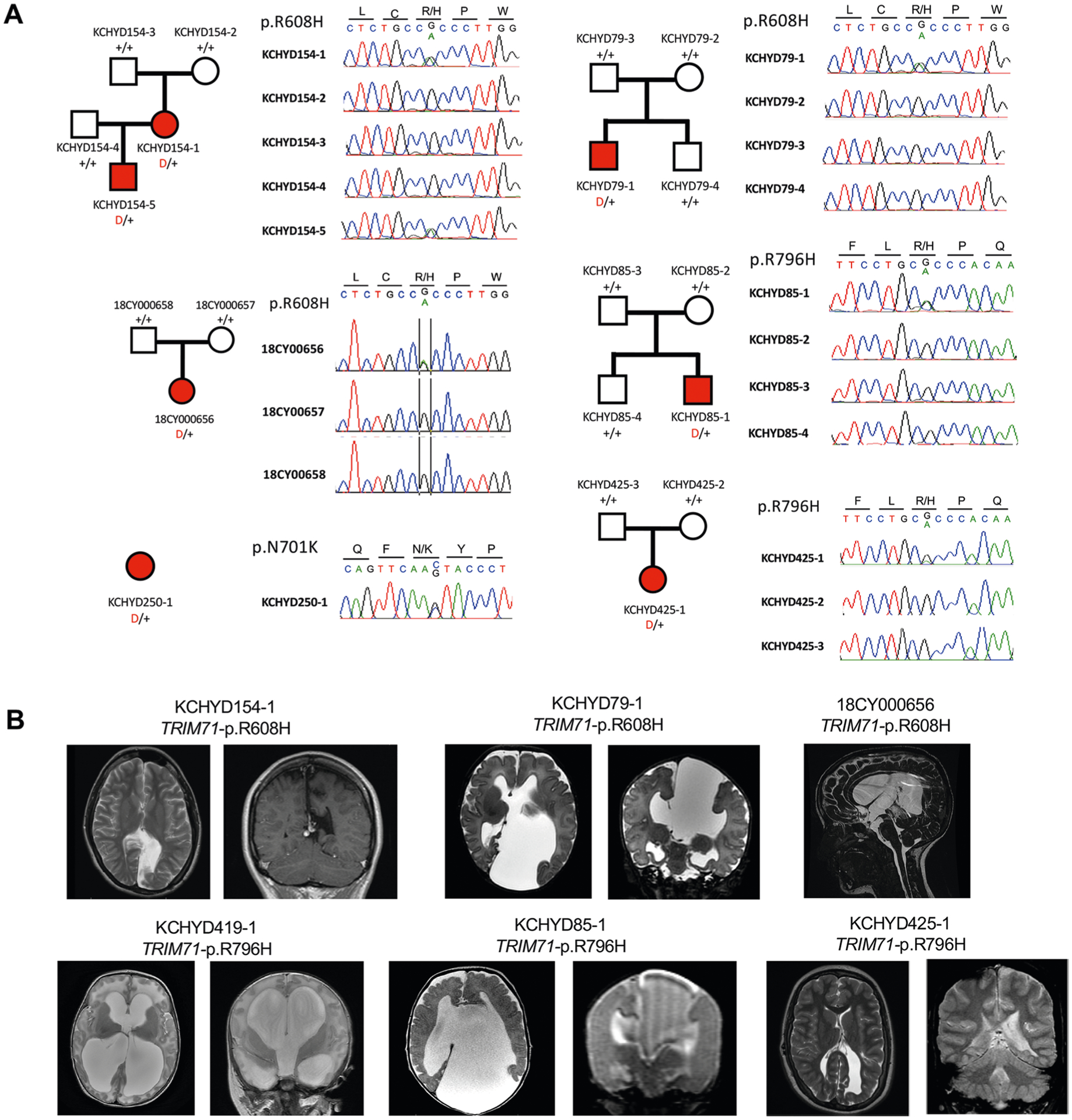
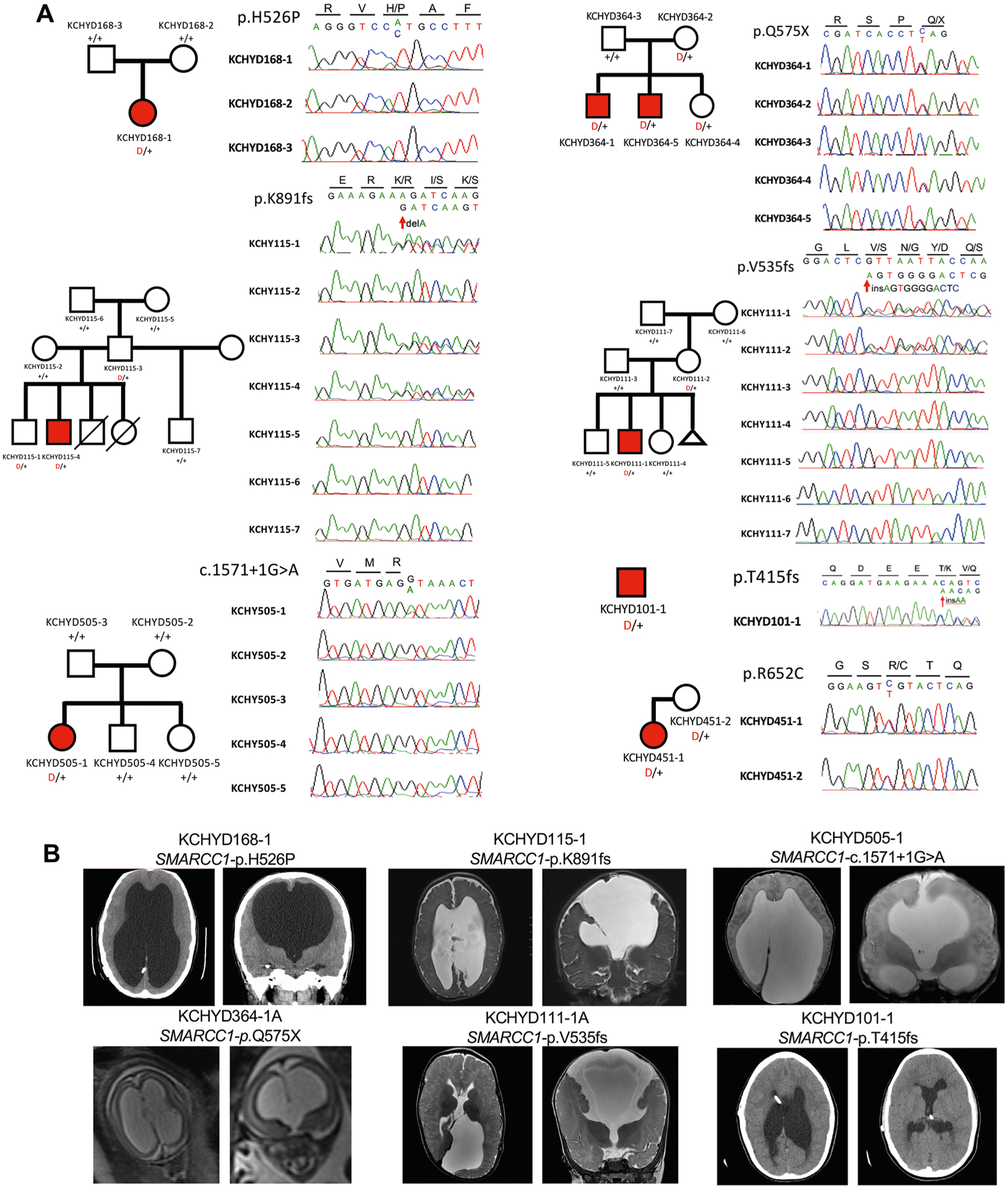
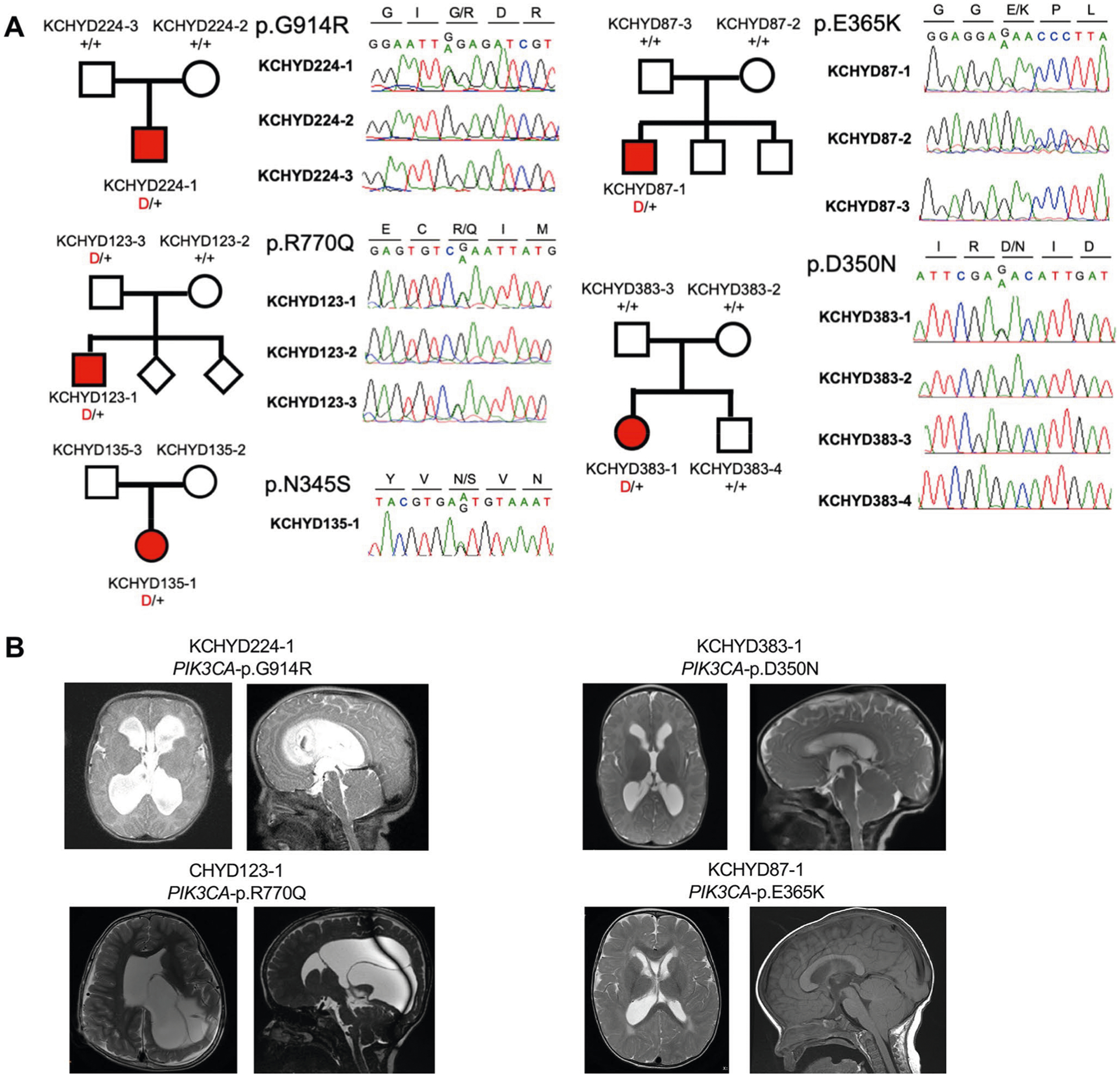
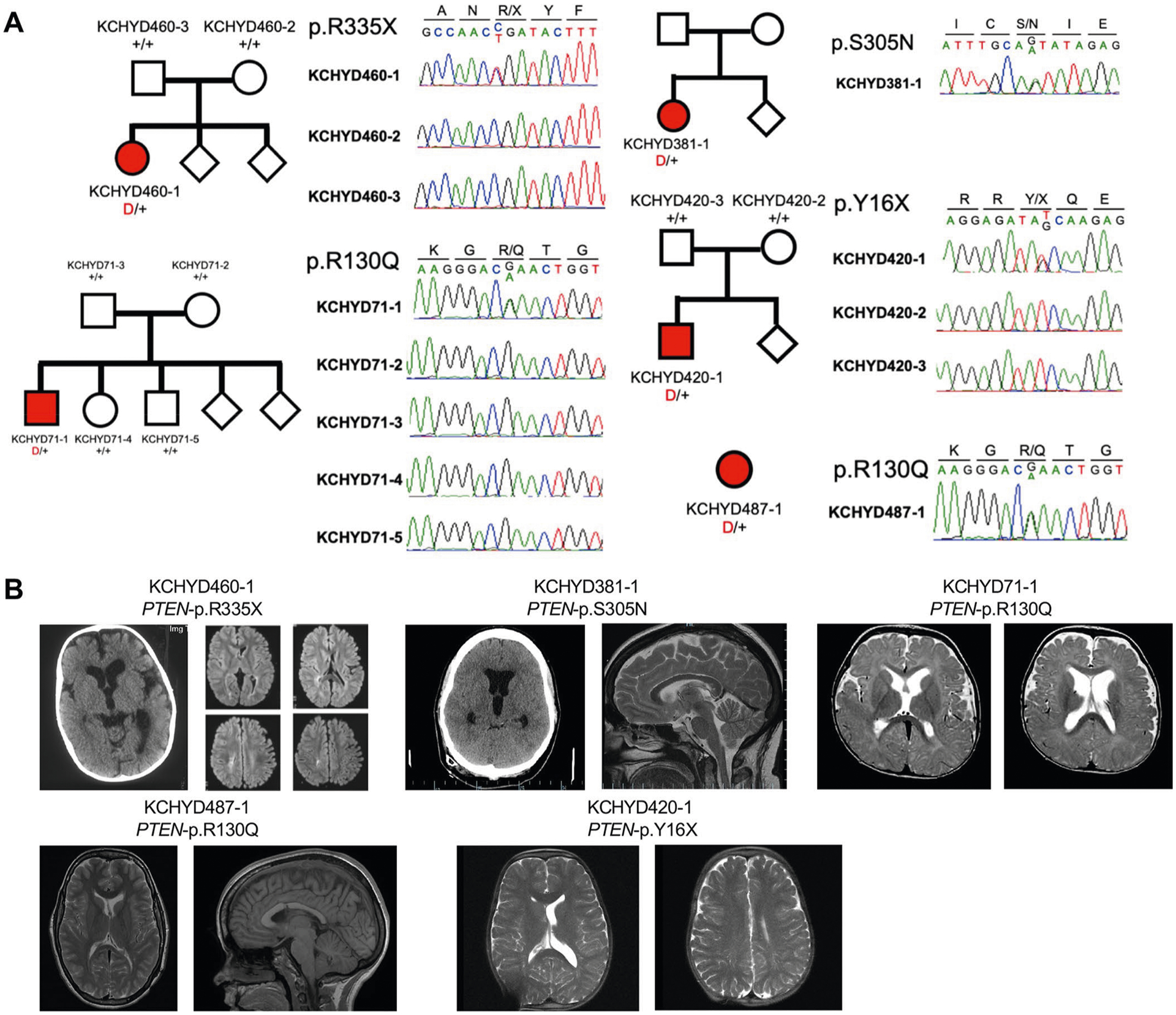
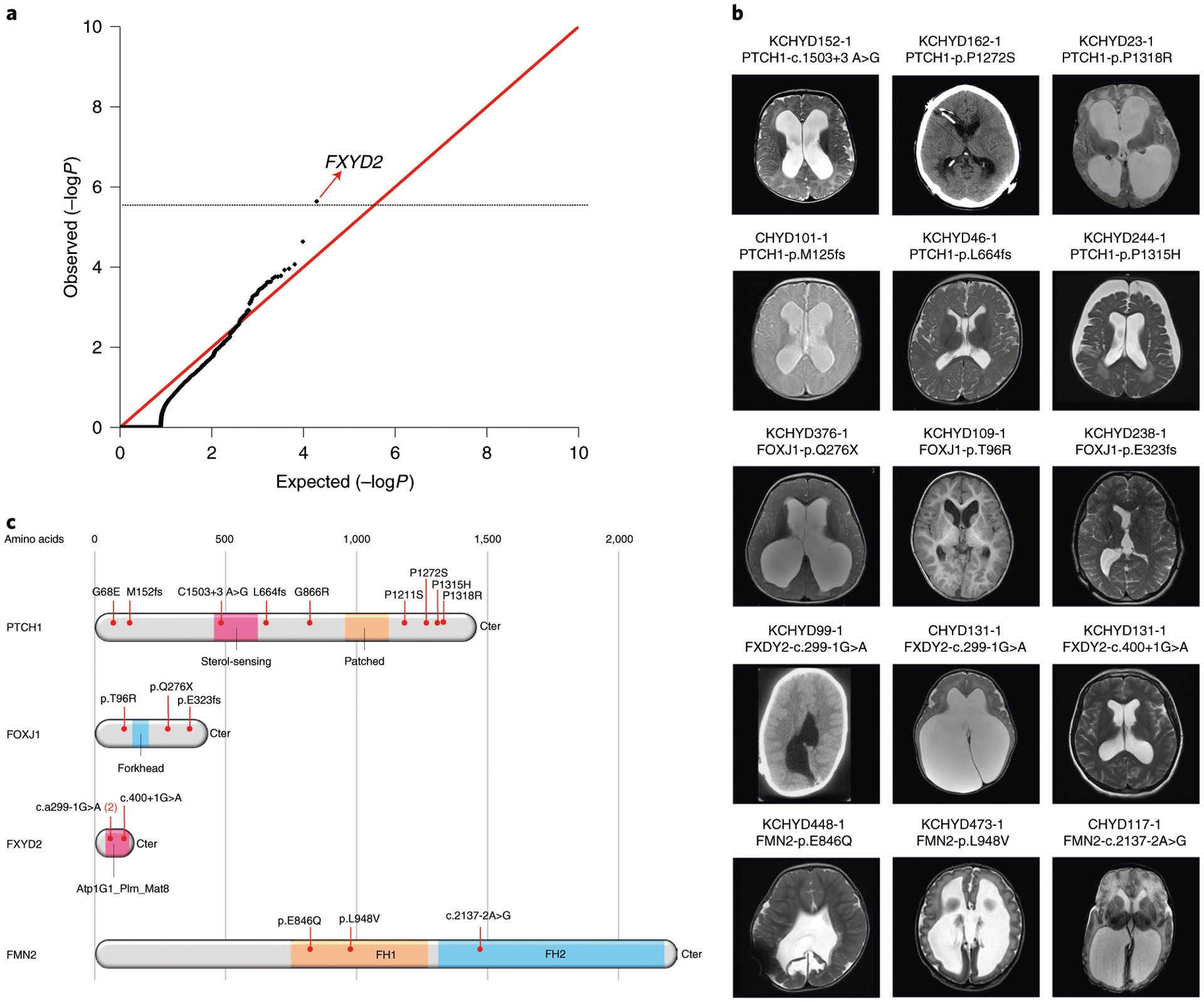
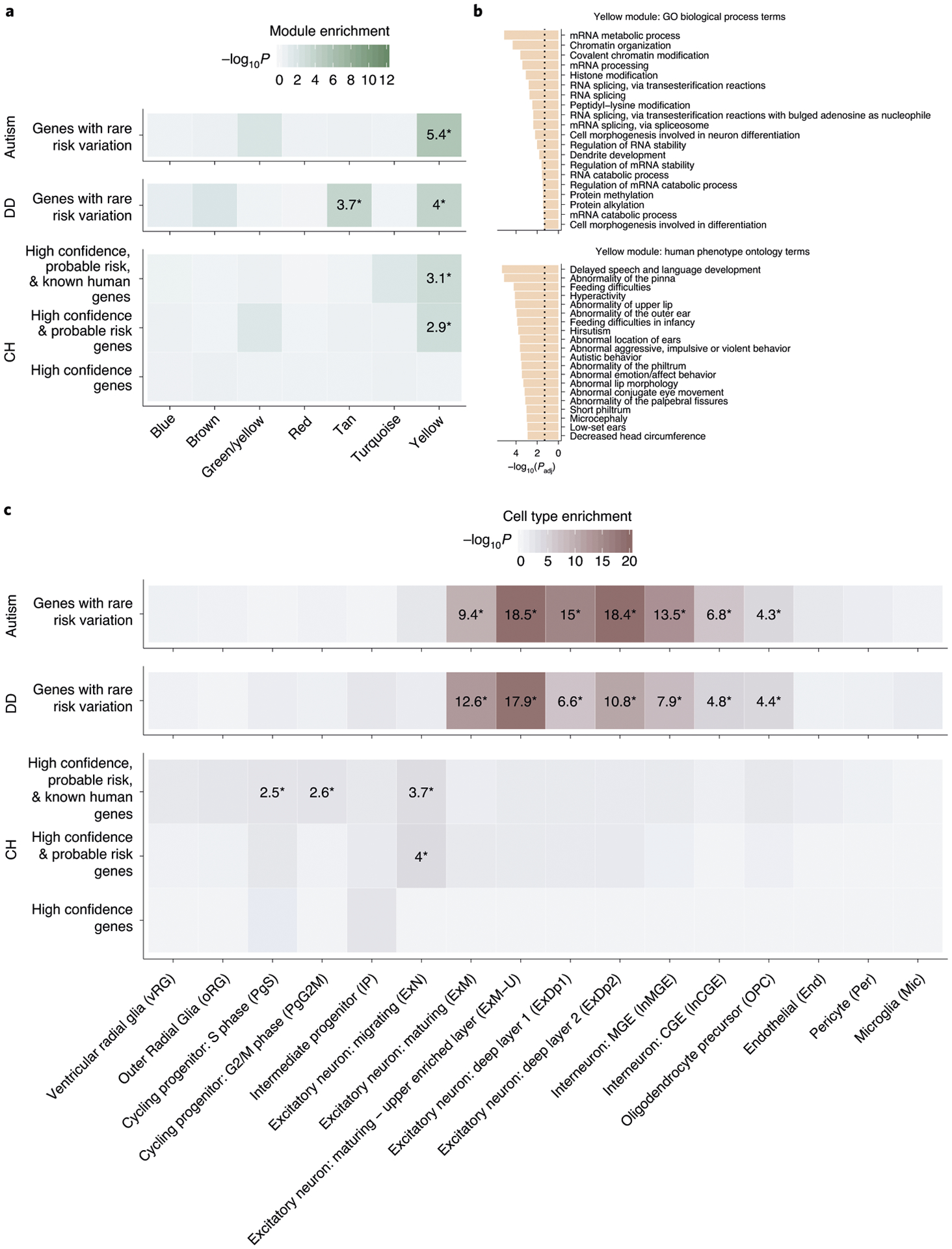
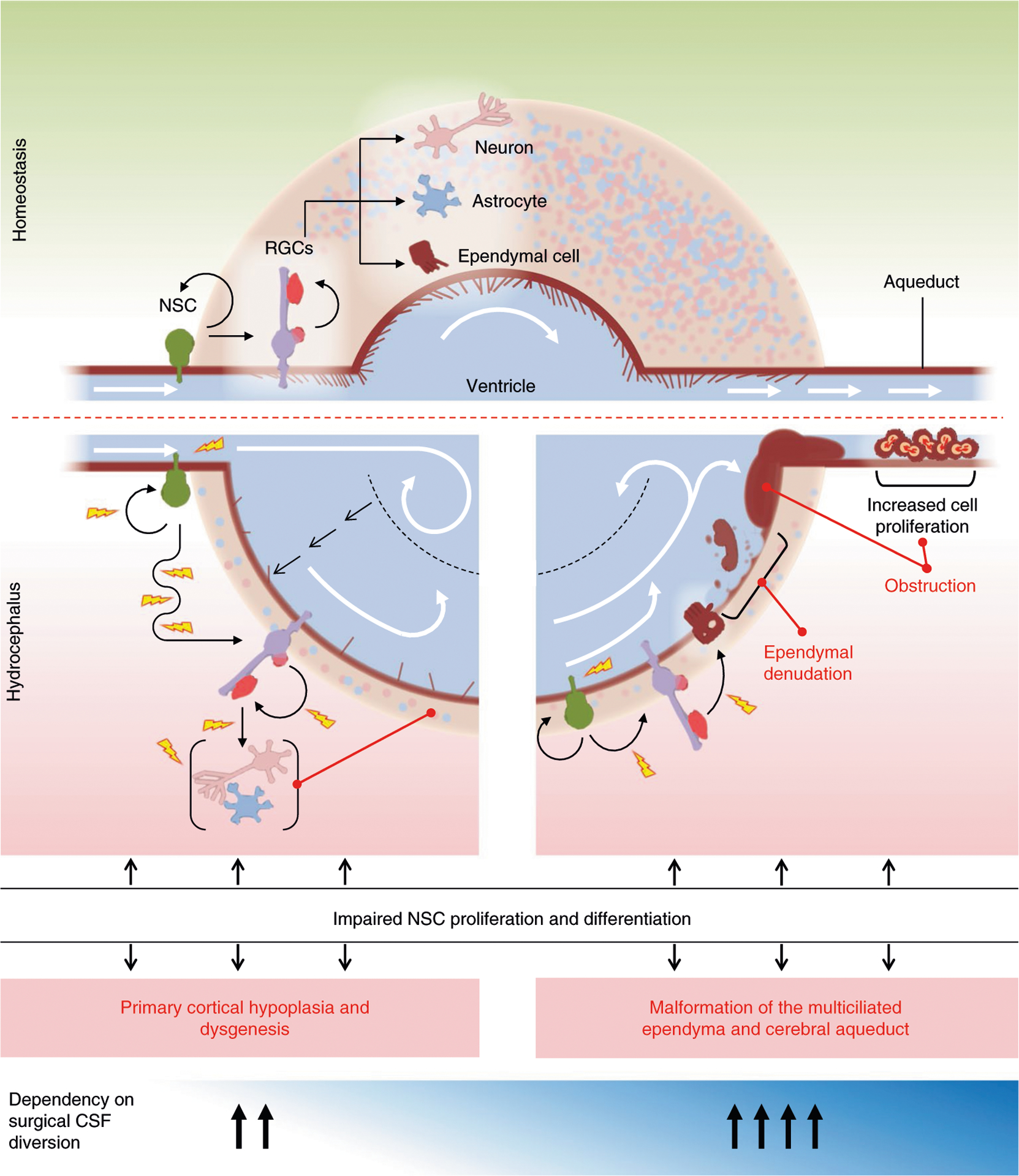
Comment in
-
Unlocking the genetic complexity of congenital hydrocephalus.Nat Med. 2020 Nov;26(11):1682-1683. doi: 10.1038/s41591-020-1120-0. Nat Med. 2020. PMID: 33106667 No abstract available.
References
-
- Albright AL, Adelson PD & Pollack IF Principles and Practice of Pediatric Neurosurgery (Thieme, 2008).
-
- Bondurant CP & Jimenez DF Epidemiology of cerebrospinal fluid shunting. Pediatr. Neurosurg 23, 254–258 (1995). - PubMed
-
- Lindquist B, Carlsson G, Persson EK & Uvebrant P Behavioural problems and autism in children with hydrocephalus: a population-based study. Eur. Child Adolesc. Psychiatry 15, 214–219 (2006). - PubMed
-
- Kahle KT, Kulkarni AV, Limbrick DD Jr. & Warf BC Hydrocephalus in children. Lancet 387, 788–799 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 NS109358/NS/NINDS NIH HHS/United States
- R01 AI145057/AI/NIAID NIH HHS/United States
- R01 NS111029/NS/NINDS NIH HHS/United States
- DP1 HD086071/HD/NICHD NIH HHS/United States
- R00 HL143036/HL/NHLBI NIH HHS/United States
- U54 HG006504/HG/NHGRI NIH HHS/United States
- K99 HL143036/HL/NHLBI NIH HHS/United States
- F31 NS115519/NS/NINDS NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- T32 HD007149/HD/NICHD NIH HHS/United States
- T32 GM007205/GM/NIGMS NIH HHS/United States
- TL1 TR001864/TR/NCATS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
