

Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications
- PMID: 33079175
- PMCID: PMC7609030
- DOI: 10.1042/BST20191091
Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications
Abstract
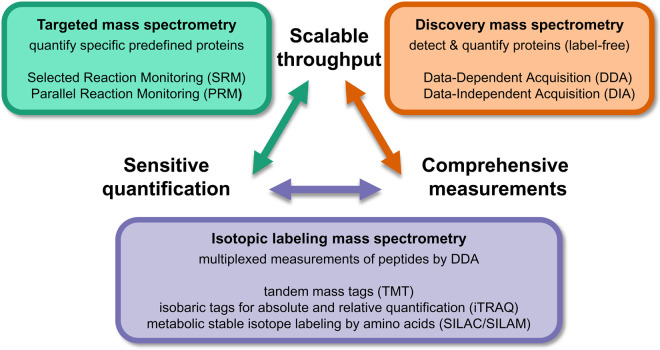
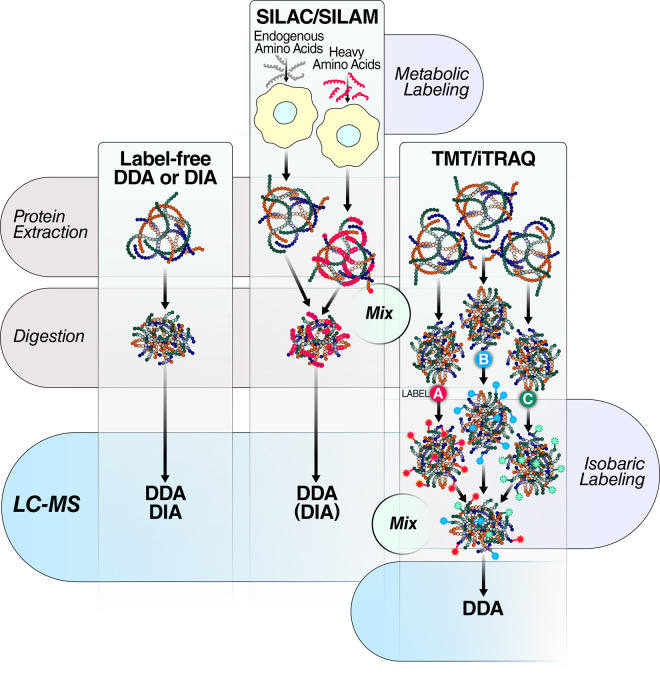
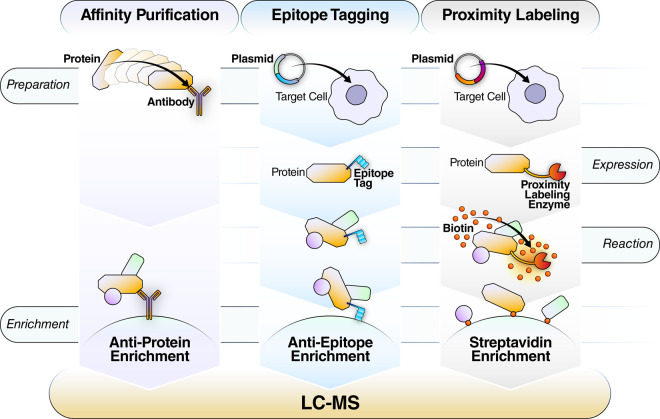
Research into the basic biology of human health and disease, as well as translational human research and clinical applications, all benefit from the growing accessibility and versatility of mass spectrometry (MS)-based proteomics. Although once limited in throughput and sensitivity, proteomic studies have quickly grown in scope and scale over the last decade due to significant advances in instrumentation, computational approaches, and bio-sample preparation. Here, we review these latest developments in MS and highlight how these techniques are used to study the mechanisms, diagnosis, and treatment of human diseases. We first describe recent groundbreaking technological advancements for MS-based proteomics, including novel data acquisition techniques and protein quantification approaches. Next, we describe innovations that enable the unprecedented depth of coverage in protein signaling and spatiotemporal protein distributions, including studies of post-translational modifications, protein turnover, and single-cell proteomics. Finally, we explore new workflows to investigate protein complexes and structures, and we present new approaches for protein-protein interaction studies and intact protein or top-down MS. While these approaches are only recently incipient, we anticipate that their use in biomedical MS proteomics research will offer actionable discoveries for the improvement of human health.
Keywords: mass spectrometry; proteomics; technology.
© 2020 The Author(s).
Conflict of interest statement
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Figures
Similar articles
-
Next-generation proteomics: towards an integrative view of proteome dynamics.Nat Rev Genet. 2013 Jan;14(1):35-48. doi: 10.1038/nrg3356. Epub 2012 Dec 4. Nat Rev Genet. 2013. PMID: 23207911 Review.
-
Application of targeted mass spectrometry in bottom-up proteomics for systems biology research.J Proteomics. 2018 Oct 30;189:75-90. doi: 10.1016/j.jprot.2018.02.008. Epub 2018 Feb 13. J Proteomics. 2018. PMID: 29452276 Free PMC article. Review.
-
Novel Strategies to Address the Challenges in Top-Down Proteomics.J Am Soc Mass Spectrom. 2021 Jun 2;32(6):1278-1294. doi: 10.1021/jasms.1c00099. Epub 2021 May 13. J Am Soc Mass Spectrom. 2021. PMID: 33983025 Free PMC article. Review.
-
Advancements in mass spectrometry-based proteomics: a new era in pathology research and diagnostics.Pathologie (Heidelb). 2024 Nov;45(Suppl 1):56-62. doi: 10.1007/s00292-024-01390-x. Epub 2024 Nov 7. Pathologie (Heidelb). 2024. PMID: 39508868 Review. English.
-
Mass-spectrometry-based proteomics: from single cells to clinical applications.Nature. 2025 Feb;638(8052):901-911. doi: 10.1038/s41586-025-08584-0. Epub 2025 Feb 26. Nature. 2025. PMID: 40011722 Review.
Cited by
-
Proximity labeling and other novel mass spectrometric approaches for spatiotemporal protein dynamics.Expert Rev Proteomics. 2021 Sep;18(9):757-765. doi: 10.1080/14789450.2021.1976149. Epub 2021 Sep 15. Expert Rev Proteomics. 2021. PMID: 34496693 Free PMC article. Review.
-
Improved SILAC Quantification with Data-Independent Acquisition to Investigate Bortezomib-Induced Protein Degradation.J Proteome Res. 2021 Apr 2;20(4):1918-1927. doi: 10.1021/acs.jproteome.0c00938. Epub 2021 Mar 25. J Proteome Res. 2021. PMID: 33764077 Free PMC article.
-
Locality-sensitive hashing enables efficient and scalable signal classification in high-throughput mass spectrometry raw data.BMC Bioinformatics. 2022 Jul 20;23(1):287. doi: 10.1186/s12859-022-04833-5. BMC Bioinformatics. 2022. PMID: 35858828 Free PMC article.
-
Rustims: An Open-Source Framework for Rapid Development and Processing of timsTOF Data-Dependent Acquisition Data.J Proteome Res. 2025 May 2;24(5):2358-2368. doi: 10.1021/acs.jproteome.4c00966. Epub 2025 Apr 22. J Proteome Res. 2025. PMID: 40260647 Free PMC article.
-
Ten questions to AI regarding the present and future of proteomics.Front Mol Biosci. 2023 Nov 23;10:1295721. doi: 10.3389/fmolb.2023.1295721. eCollection 2023. Front Mol Biosci. 2023. PMID: 38074090 Free PMC article.
References
-
- Schilling B., Rardin M.J., MacLean B.X., Zawadzka A.M., Frewen B.E., Cusack M.P. et al. (2012) Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation. Mol. Cell. Proteomics 11, 202–214 10.1074/mcp.M112.017707 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources