

Guanosine triphosphate links MYC-dependent metabolic and ribosome programs in small-cell lung cancer
- PMID: 33079728
- PMCID: PMC7773395
- DOI: 10.1172/JCI139929
Guanosine triphosphate links MYC-dependent metabolic and ribosome programs in small-cell lung cancer
Abstract
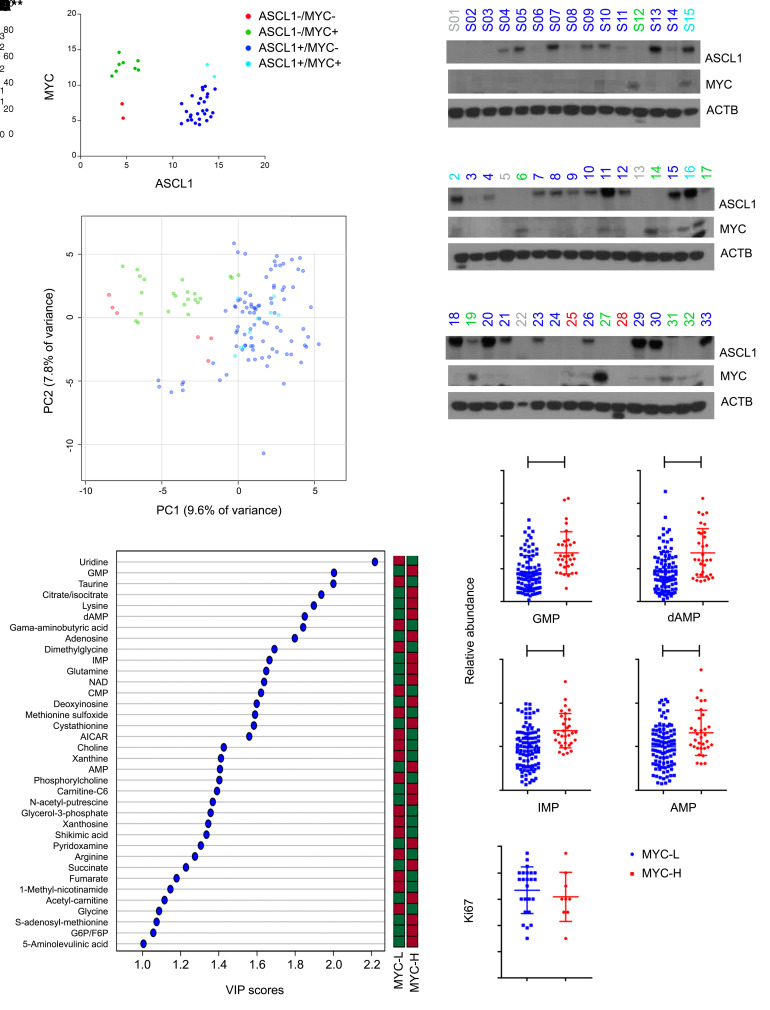
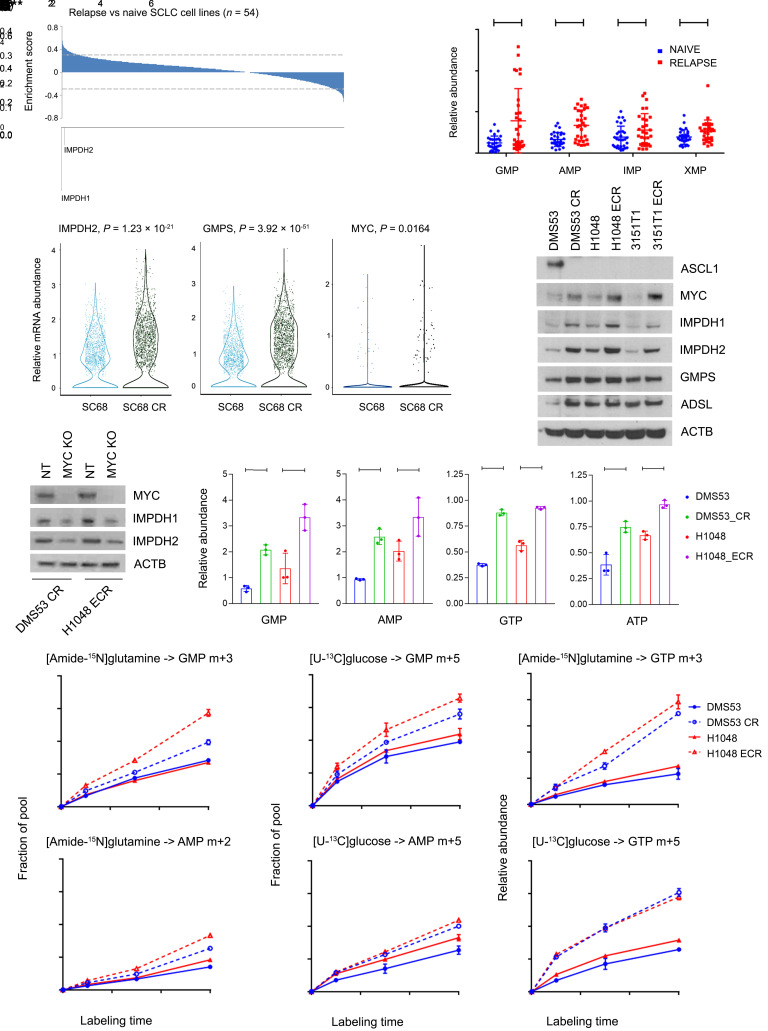
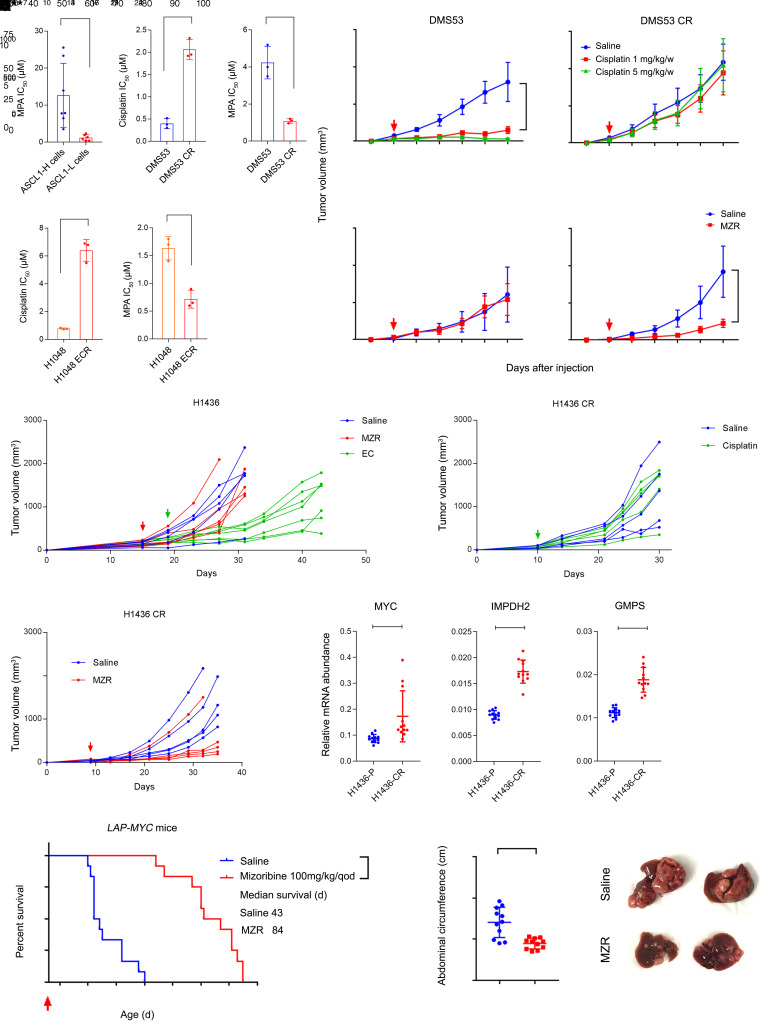
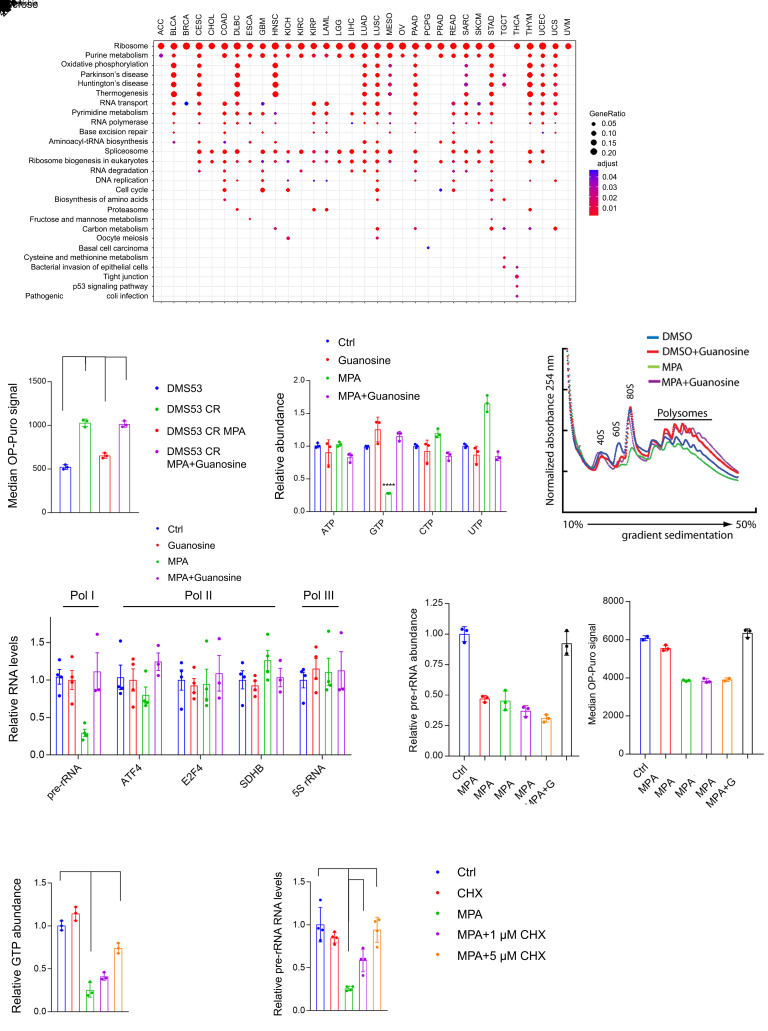
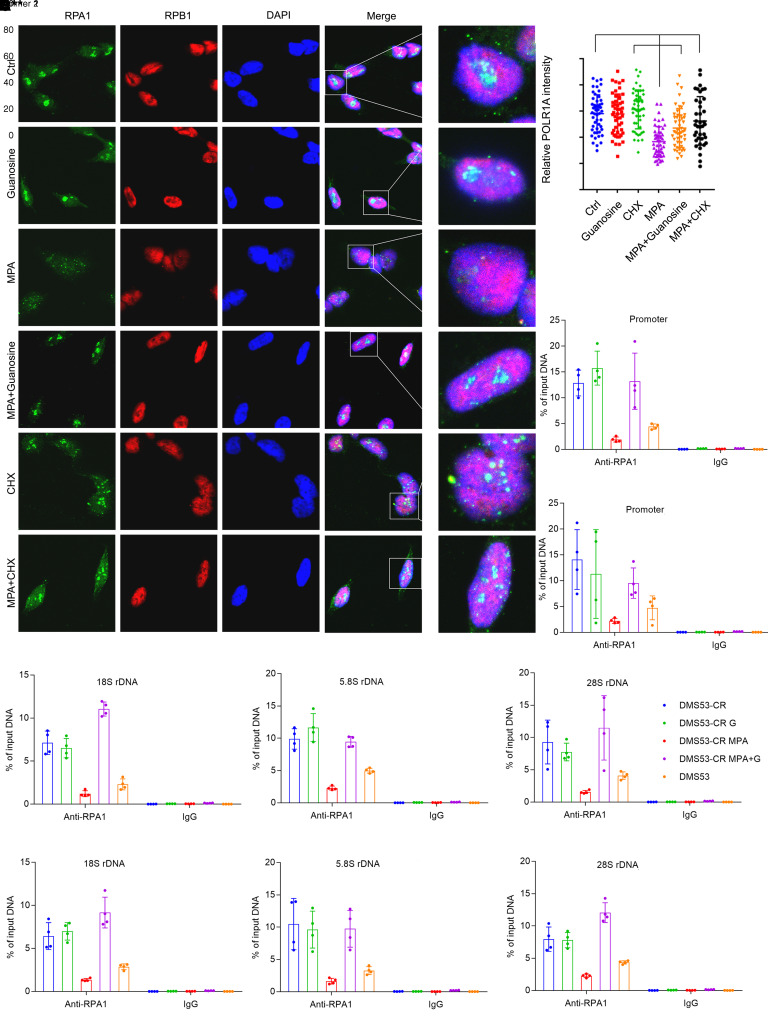
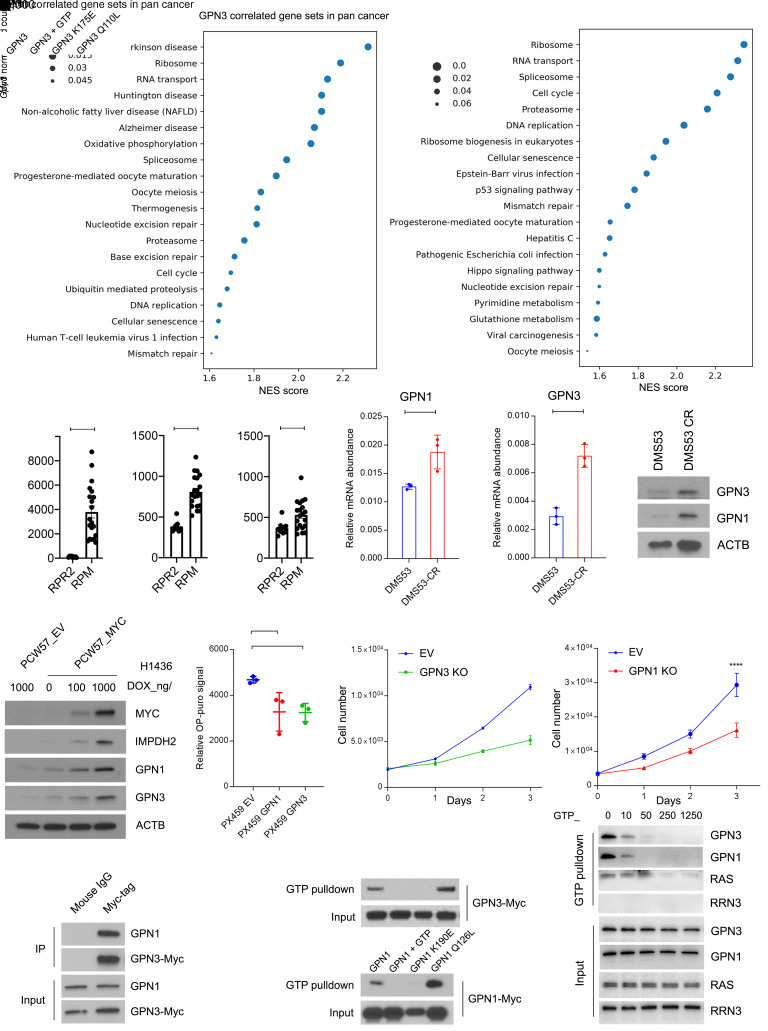
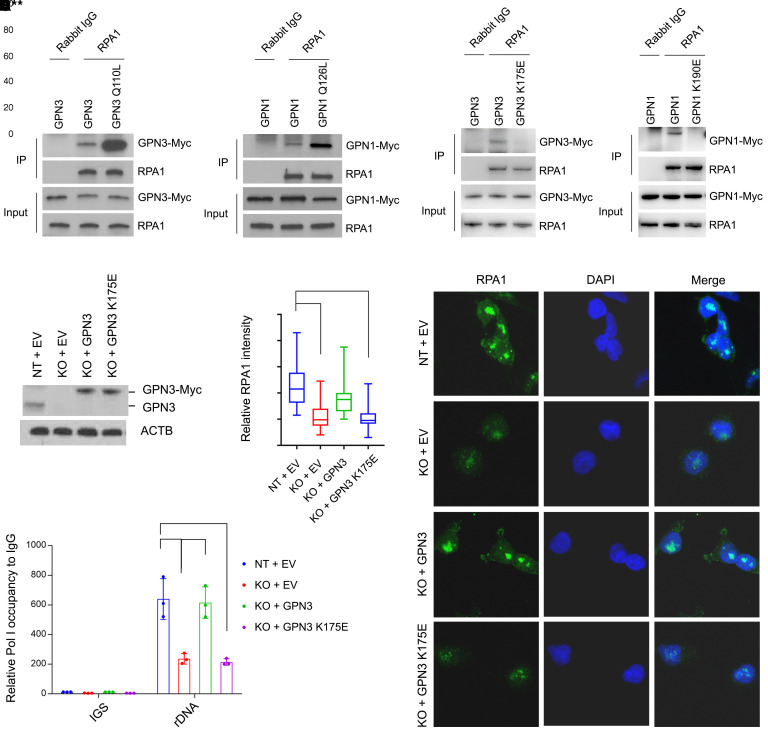
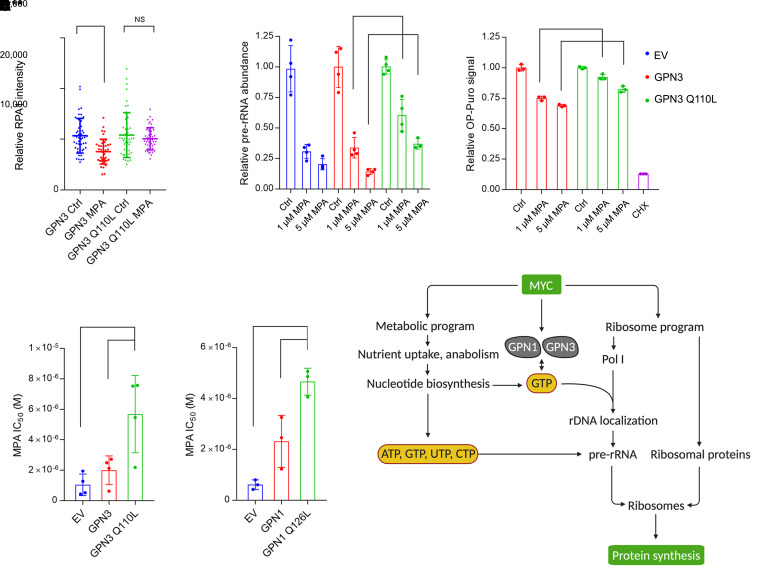
MYC stimulates both metabolism and protein synthesis, but how cells coordinate these complementary programs is unknown. Previous work reported that, in a subset of small-cell lung cancer (SCLC) cell lines, MYC activates guanosine triphosphate (GTP) synthesis and results in sensitivity to inhibitors of the GTP synthesis enzyme inosine monophosphate dehydrogenase (IMPDH). Here, we demonstrated that primary MYChi human SCLC tumors also contained abundant guanosine nucleotides. We also found that elevated MYC in SCLCs with acquired chemoresistance rendered these otherwise recalcitrant tumors dependent on IMPDH. Unexpectedly, our data indicated that IMPDH linked the metabolic and protein synthesis outputs of oncogenic MYC. Coexpression analysis placed IMPDH within the MYC-driven ribosome program, and GTP depletion prevented RNA polymerase I (Pol I) from localizing to ribosomal DNA. Furthermore, the GTPases GPN1 and GPN3 were upregulated by MYC and directed Pol I to ribosomal DNA. Constitutively GTP-bound GPN1/3 mutants mitigated the effect of GTP depletion on Pol I, protecting chemoresistant SCLC cells from IMPDH inhibition. GTP therefore functioned as a metabolic gate tethering MYC-dependent ribosome biogenesis to nucleotide sufficiency through GPN1 and GPN3. IMPDH dependence is a targetable vulnerability in chemoresistant MYChi SCLC.
Keywords: Intermediary metabolism; Lung cancer; Metabolism; Oncogenes; Oncology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
