

Neutral components show a hierarchical community structure in the genotype-phenotype map of RNA secondary structure
- PMID: 33081646
- PMCID: PMC7653385
- DOI: 10.1098/rsif.2020.0608
Neutral components show a hierarchical community structure in the genotype-phenotype map of RNA secondary structure
Abstract
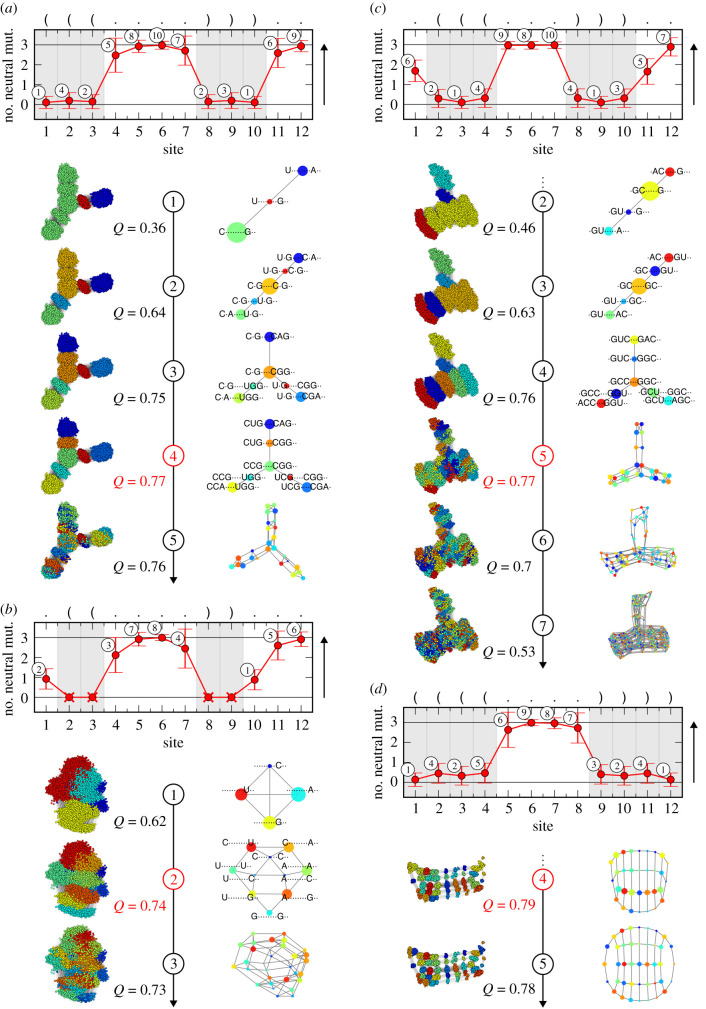
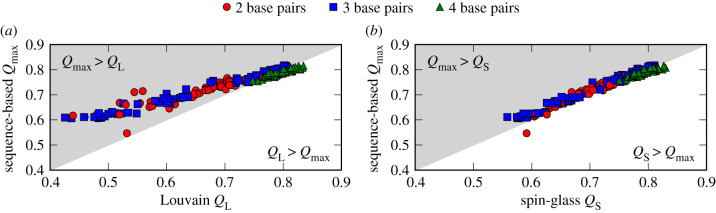
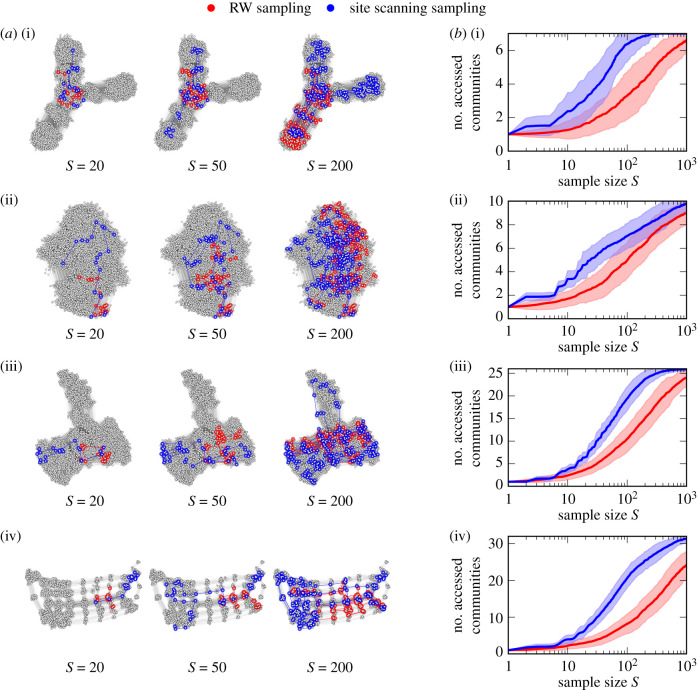
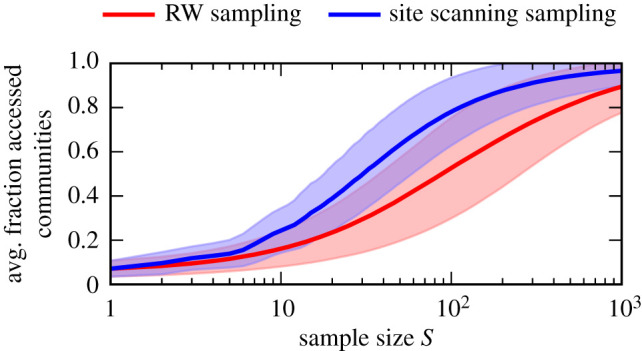
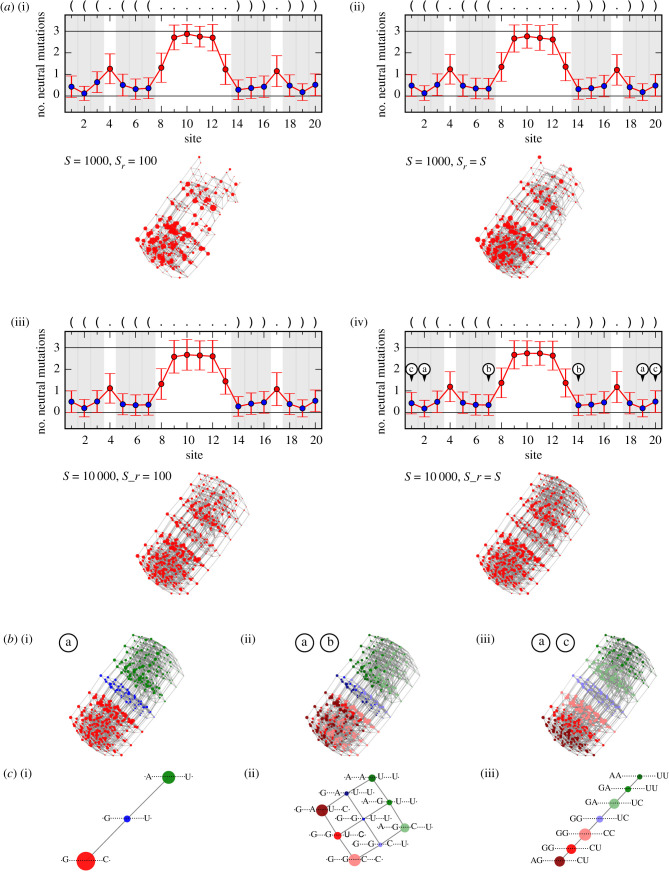
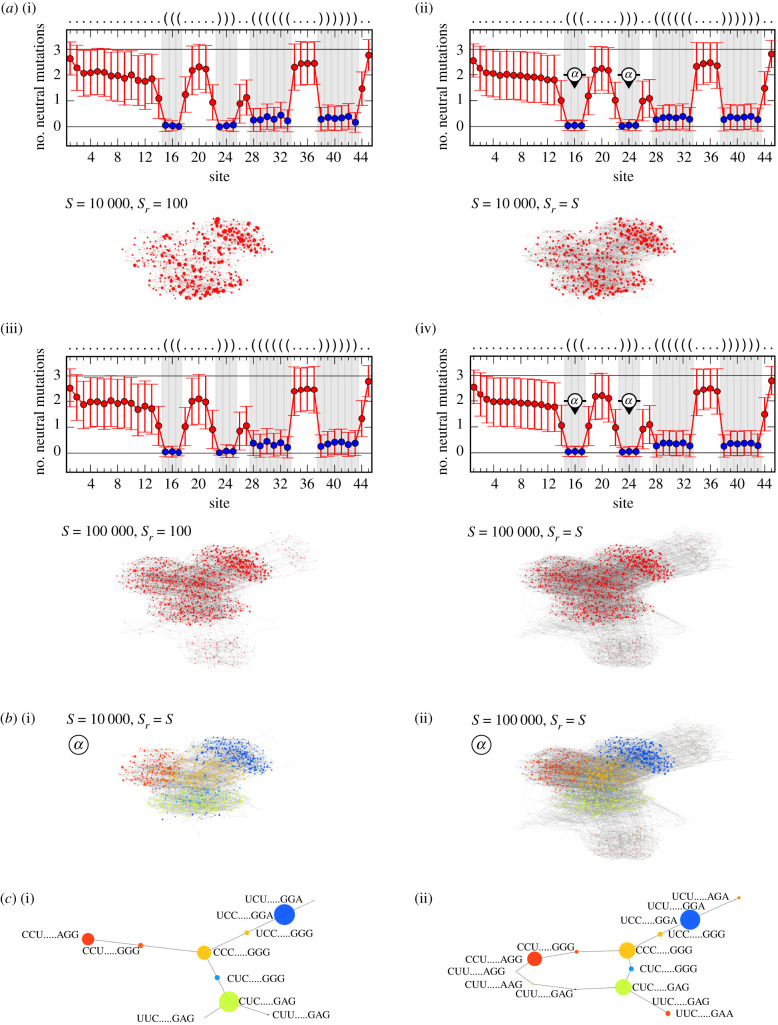
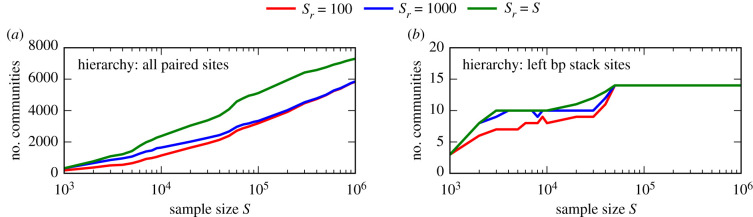
Genotype-phenotype (GP) maps describe the relationship between biological sequences and structural or functional outcomes. They can be represented as networks in which genotypes are the nodes, and one-point mutations between them are the edges. The genotypes that map to the same phenotype form subnetworks consisting of one or multiple disjoint connected components-so-called neutral components (NCs). For the GP map of RNA secondary structure, the NCs have been found to exhibit distinctive network features that can affect the dynamical processes taking place on them. Here, we focus on the community structure of RNA secondary structure NCs. Building on previous findings, we introduce a method to reveal the hierarchical community structure solely from the sequence constraints and composition of the genotypes that form a given NC. Thereby, we obtain modularity values similar to common community detection algorithms, which are much more complex. From this knowledge, we endorse a sampling method that allows a fast exploration of the different communities of a given NC. Furthermore, we introduce a way to estimate the community structure from genotype samples, which is useful when an exhaustive analysis of the NC is not feasible, as is the case for longer sequence lengths.
Keywords: RNA secondary structure; community structure; genotype–phenotype map; network.
Conflict of interest statement
We declare we have no competing interests.
Figures
Similar articles
-
Using small samples to estimate neutral component size and robustness in the genotype-phenotype map of RNA secondary structure.J R Soc Interface. 2020 May;17(166):20190784. doi: 10.1098/rsif.2019.0784. Epub 2020 May 20. J R Soc Interface. 2020. PMID: 32429824 Free PMC article.
-
Phenotypes can be robust and evolvable if mutations have non-local effects on sequence constraints.J R Soc Interface. 2018 Jan;15(138):20170618. doi: 10.1098/rsif.2017.0618. J R Soc Interface. 2018. PMID: 29321270 Free PMC article.
-
Epistasis can lead to fragmented neutral spaces and contingency in evolution.Proc Biol Sci. 2012 May 7;279(1734):1777-83. doi: 10.1098/rspb.2011.2183. Epub 2011 Dec 7. Proc Biol Sci. 2012. PMID: 22158953 Free PMC article.
-
Structural properties of genotype-phenotype maps.J R Soc Interface. 2017 Jul;14(132):20170275. doi: 10.1098/rsif.2017.0275. J R Soc Interface. 2017. PMID: 28679667 Free PMC article. Review.
-
Evolution in silico and in vitro: the RNA model.Biol Chem. 2001 Sep;382(9):1301-14. doi: 10.1515/BC.2001.162. Biol Chem. 2001. PMID: 11688713 Review.
Cited by
-
Insertions and deletions in the RNA sequence-structure map.J R Soc Interface. 2021 Oct;18(183):20210380. doi: 10.1098/rsif.2021.0380. Epub 2021 Oct 6. J R Soc Interface. 2021. PMID: 34610259 Free PMC article.
-
Maximum mutational robustness in genotype-phenotype maps follows a self-similar blancmange-like curve.J R Soc Interface. 2023 Jul;20(204):20230169. doi: 10.1098/rsif.2023.0169. Epub 2023 Jul 26. J R Soc Interface. 2023. PMID: 37491910 Free PMC article.
-
Probabilistic Genotype-Phenotype Maps Reveal Mutational Robustness of RNA Folding, Spin Glasses, and Quantum Circuits.ArXiv [Preprint]. 2025 Jan 3:arXiv:2301.01847v3. ArXiv. 2025. PMID: 36713233 Free PMC article. Preprint.
-
Non-Poissonian Bursts in the Arrival of Phenotypic Variation Can Strongly Affect the Dynamics of Adaptation.Mol Biol Evol. 2024 Jun 1;41(6):msae085. doi: 10.1093/molbev/msae085. Mol Biol Evol. 2024. PMID: 38693911 Free PMC article.
References
-
- Wright S. 1932. The roles of mutation, inbreeding, crossbreeding, and selection in evolution. Proc. Sixth Int. Congr. Genet. 1, 355–366.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
