

Porcine circovirus 2 (PCV2) population study in experimentally infected pigs developing PCV2-systemic disease or a subclinical infection
- PMID: 33082419
- PMCID: PMC7576782
- DOI: 10.1038/s41598-020-74627-3
Porcine circovirus 2 (PCV2) population study in experimentally infected pigs developing PCV2-systemic disease or a subclinical infection
Abstract
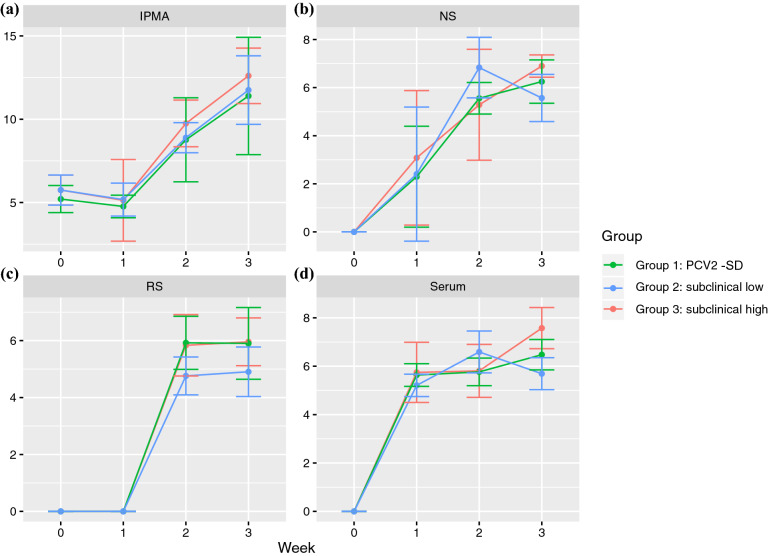
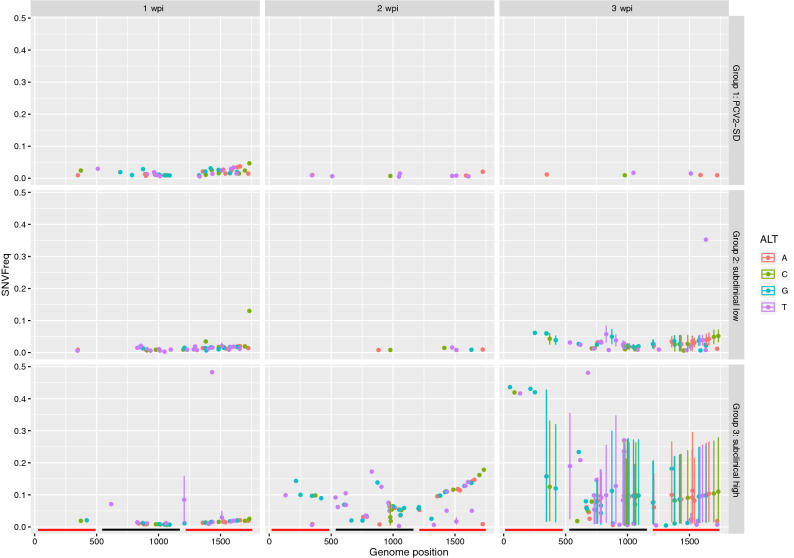
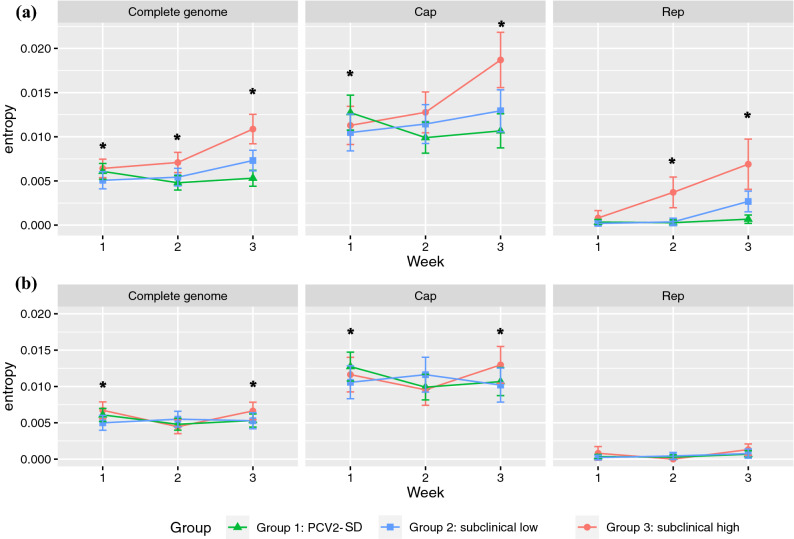
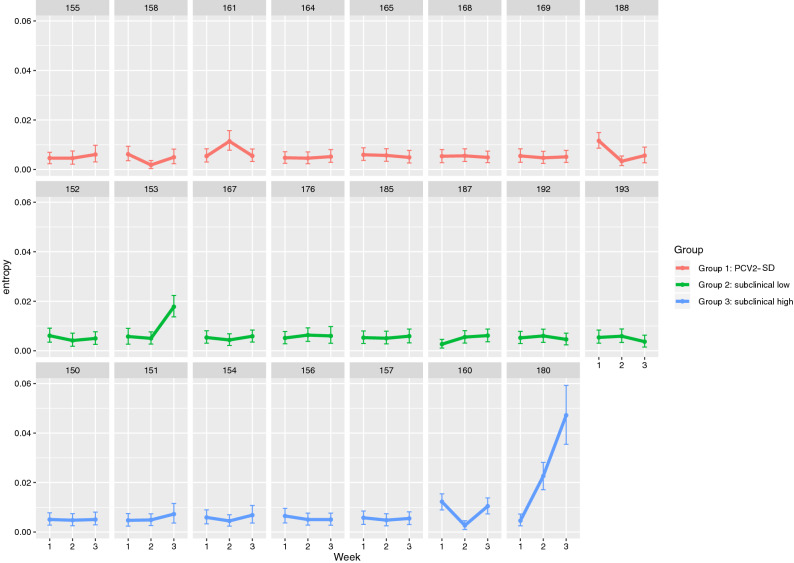
Porcine circovirus 2 (PCV2) is a single stranded DNA virus with one of the highest mutation rates among DNA viruses. This ability allows it to generate a cloud of mutants constantly providing new opportunities to adapt and evade the immune system. This pig pathogen is associated to many diseases, globally called porcine circovirus diseases (PCVD) and has been a threat to pig industry since its discovery in the early 90's. Although 11 ORFs have been predicted from its genome, only two main proteins have been deeply characterized, i.e. Rep and Cap. The structural Cap protein possesses the majority of the epitopic determinants of this non-enveloped virus. The evolution of PCV2 is affected by both natural and vaccine-induced immune responses, which enhances the genetic variability, especially in the most immunogenic Cap region. Intra-host variability has been also demonstrated in infected animals where long-lasting infections can take place. However, the association between this intra-host variability and pathogenesis has never been studied for this virus. Here, the within-host PCV2 variability was monitored over time by next generation sequencing during an experimental infection, demonstrating the presence of large heterogeneity. Remarkably, the level of quasispecies diversity, affecting particularly the Cap coding region, was statistically different depending on viremia levels and clinical signs detected after infection. Moreover, we proved the existence of hyper mutant subjects harboring a remarkably higher number of genetic variants. Altogether, these results suggest an interaction between genetic diversity, host immune system and disease severity.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
