

Disease-Specific Autoantibodies Induce Trained Immunity in RA Synovial Tissues and Its Gene Signature Correlates with the Response to Clinical Therapy
- PMID: 33082707
- PMCID: PMC7558774
- DOI: 10.1155/2020/2109325
Disease-Specific Autoantibodies Induce Trained Immunity in RA Synovial Tissues and Its Gene Signature Correlates with the Response to Clinical Therapy
Abstract
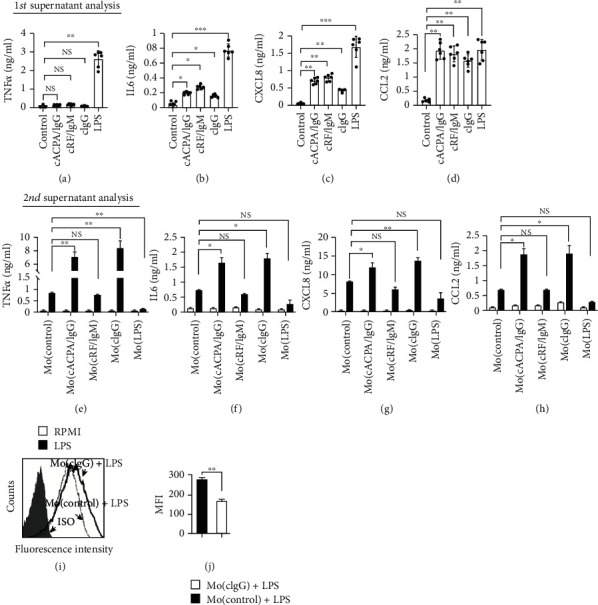
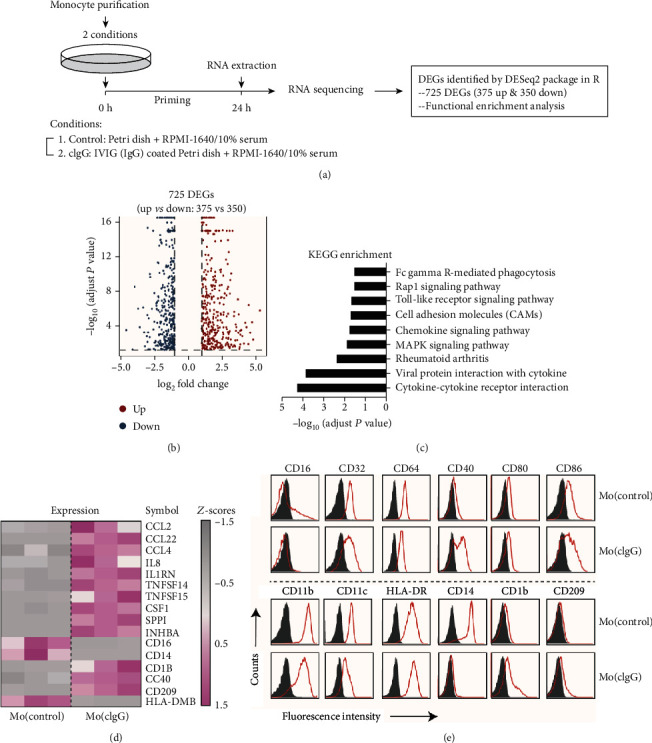
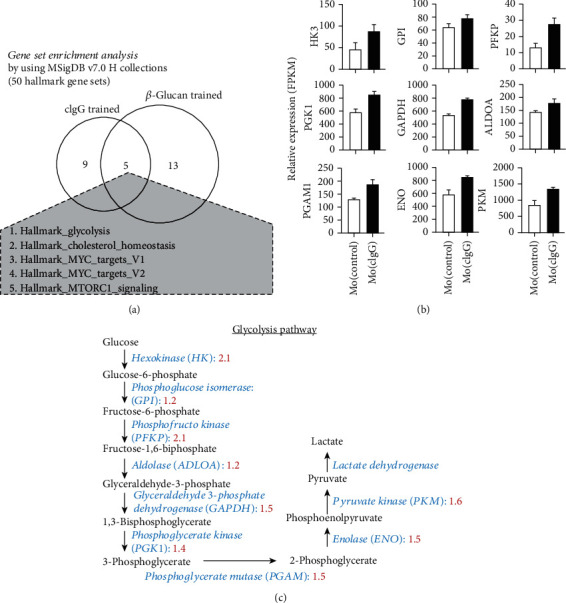
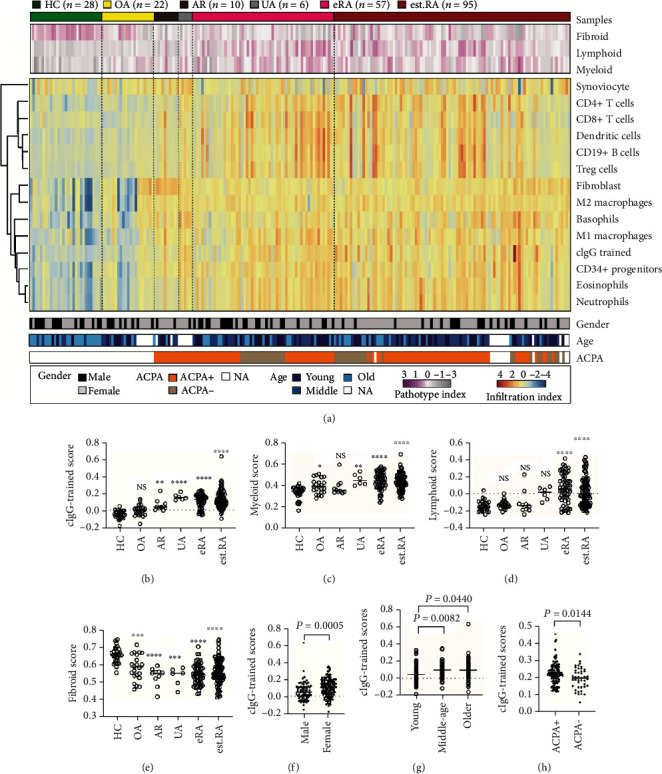
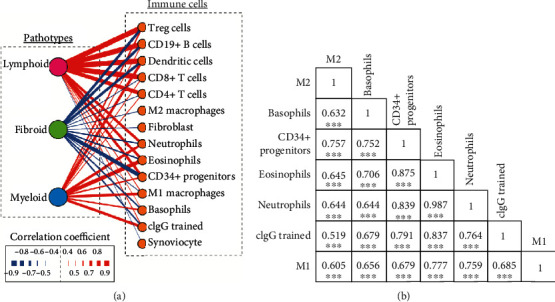
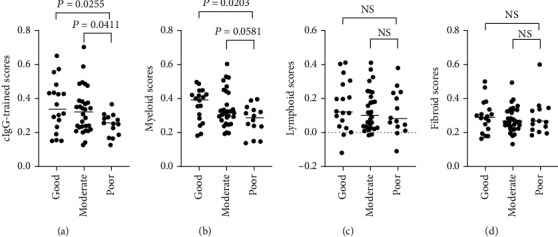
Much evidence suggests that trained immunity is inappropriately activated in the synovial tissue in rheumatoid arthritis (RA), but the underlying mechanism remains unclear. Here, we describe how RA-specific autoantibody deposits can train human monocytes to exert the hyperactive inflammatory response, particularly via the exacerbated release of tumor necrosis factor α (TNFα). Comparative transcriptomic analysis by plate-bound human IgG (cIgG) or β-glucan indicated that metabolic shift towards glycolysis is a crucial mechanism for trained immunity. Moreover, the cIgG-trained gene signatures were enriched in synovial tissues from patients with ACPA- (anticitrullinated protein antibody-) positive arthralgia and undifferentiated arthritis, and early RA and established RA bore a great resemblance to the myeloid pathotype, suggesting a historical priming event in vivo. Additionally, the expression of the cIgG-trained signatures is higher in the female, older, and ACPA-positive populations, with a predictive role in the clinical response to infliximab. We conclude that RA-specific autoantibodies can train monocytes in the inflamed lesion as early as the asymptomatic stage, which may not merely improve understanding of disease progression but may also suggest therapeutic and/or preventive strategies for autoimmune diseases.
Copyright © 2020 Xiaoli Dai et al.
Conflict of interest statement
The authors declare no conflict of interest related to this work.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
