

Optimising T cell (re)boosting strategies for adenoviral and modified vaccinia Ankara vaccine regimens in humans
- PMID: 33083029
- PMCID: PMC7550607
- DOI: 10.1038/s41541-020-00240-0
Optimising T cell (re)boosting strategies for adenoviral and modified vaccinia Ankara vaccine regimens in humans
Abstract
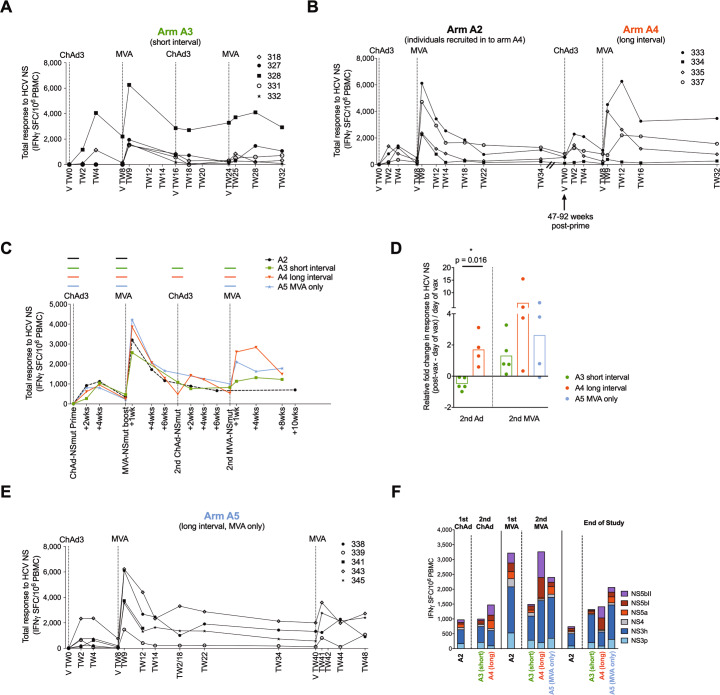
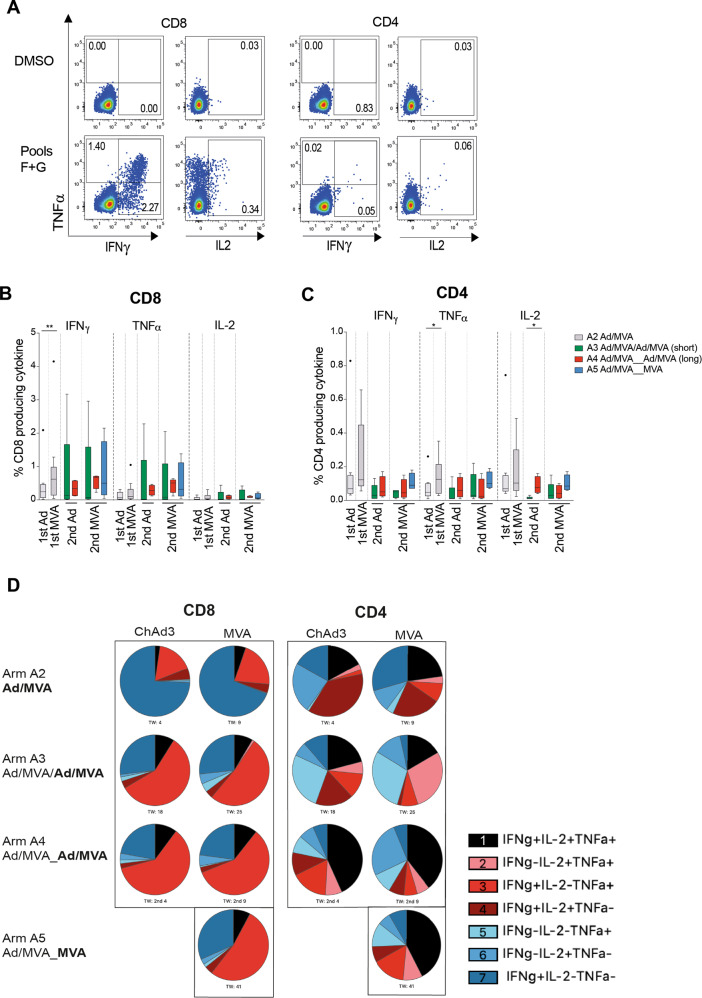
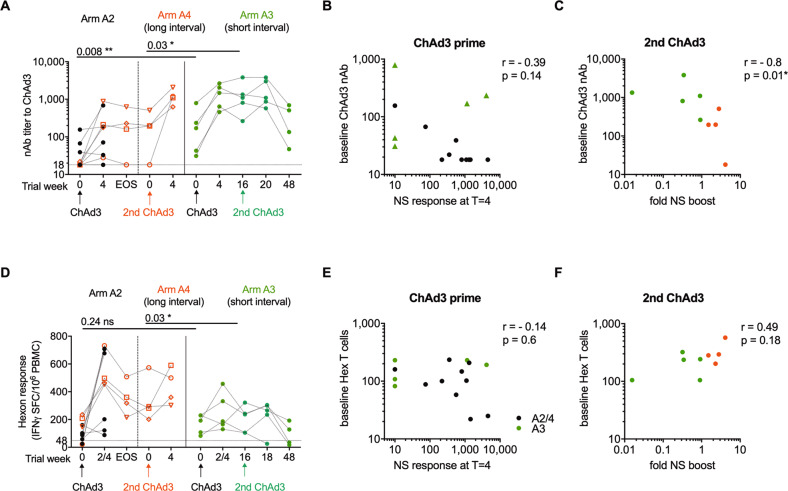
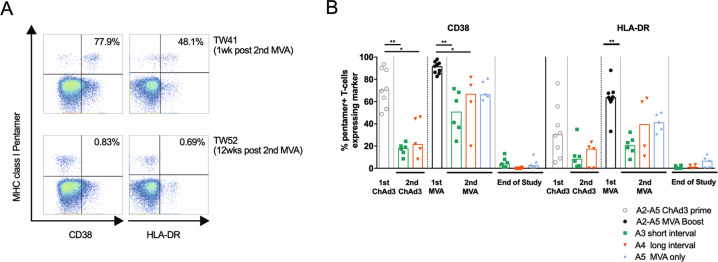
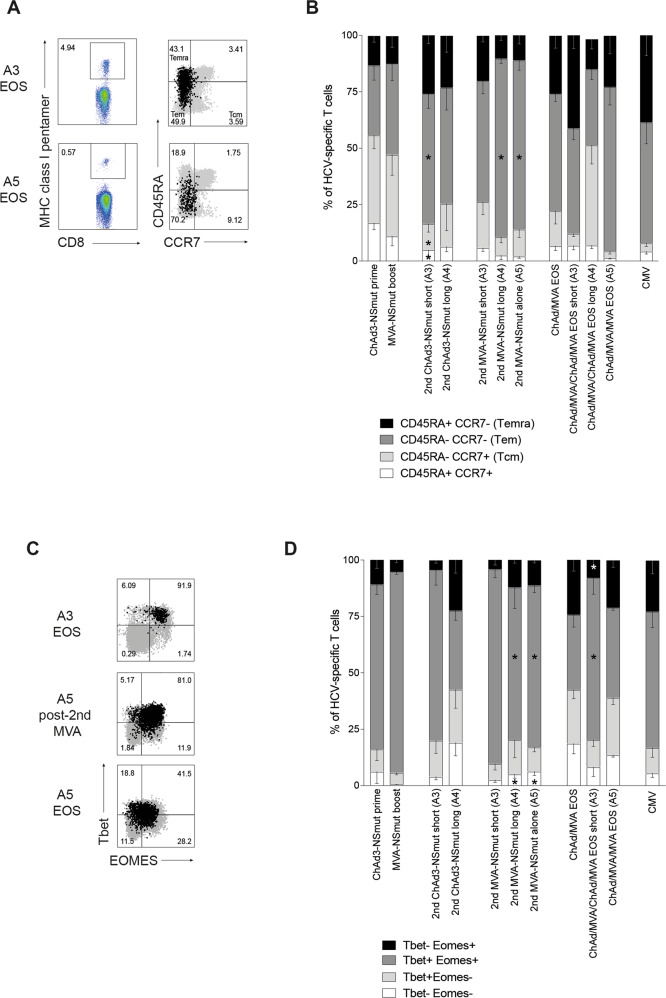
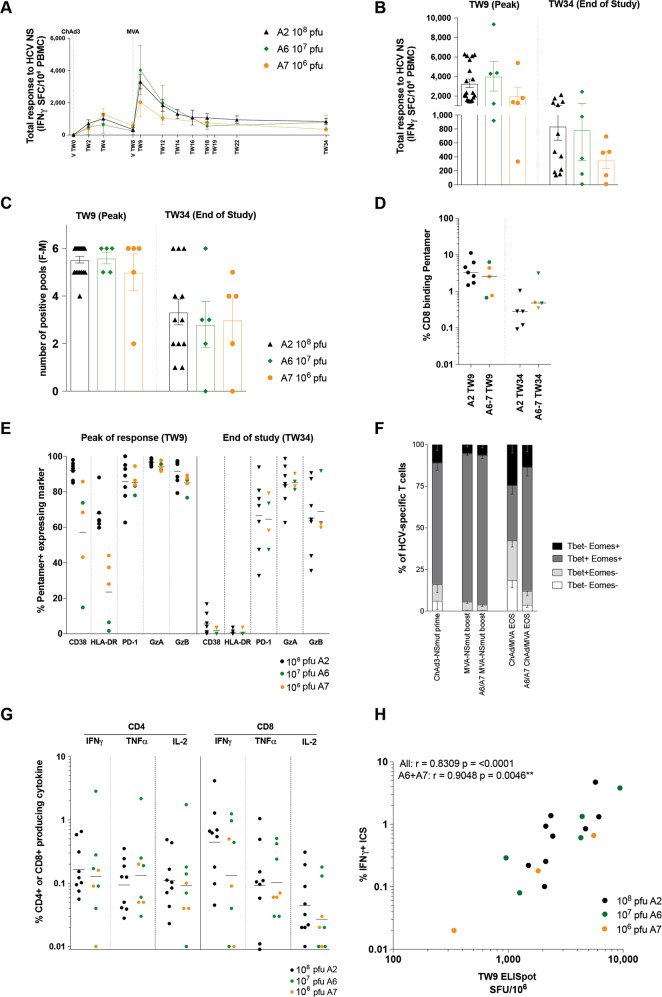
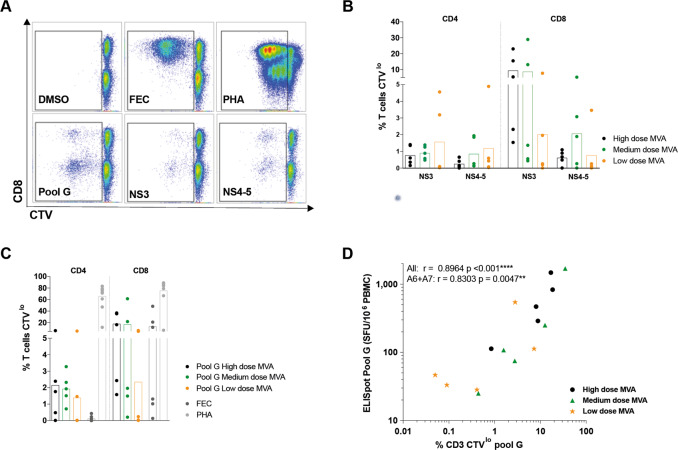
Simian adenoviral and modified vaccinia Ankara (MVA) viral vectors used in heterologous prime-boost strategies are potent inducers of T cells against encoded antigens and are in advanced testing as vaccine carriers for a wide range of infectious agents and cancers. It is unclear if these responses can be further enhanced or sustained with reboosting strategies. Furthermore, despite the challenges involved in MVA manufacture dose de-escalation has not been performed in humans. In this study, healthy volunteers received chimpanzee-derived adenovirus-3 and MVA vaccines encoding the non-structural region of hepatitis C virus (ChAd3-NSmut/MVA-NSmut) 8 weeks apart. Volunteers were then reboosted with a second round of ChAd3-NSmut/MVA-NSmut or MVA-NSmut vaccines 8 weeks or 1-year later. We also determined the capacity of reduced doses of MVA-NSmut to boost ChAd3-NSmut primed T cells. Reboosting was safe, with no enhanced reactogenicity. Reboosting after an 8-week interval led to minimal re-expansion of transgene-specific T cells. However, after a longer interval, T cell responses expanded efficiently and memory responses were enhanced. The 8-week interval regimen induced a higher percentage of terminally differentiated and effector memory T cells. Reboosting with MVA-NSmut alone was as effective as with ChAd3-NSmut/MVA-NSmut. A ten-fold lower dose of MVA (2 × 107pfu) induced high-magnitude, sustained, broad, and functional Hepatitis C virus (HCV)-specific T cell responses, equivalent to standard doses (2 × 108 pfu). Overall, we show that following Ad/MVA prime-boost vaccination reboosting is most effective after a prolonged interval and is productive with MVA alone. Importantly, we also show that a ten-fold lower dose of MVA is as potent in humans as the standard dose.
Keywords: Adaptive immunity; Live attenuated vaccines; Vaccines.
© The Author(s) 2020.
Conflict of interest statement
Competing interestsS.Co., A.F., R.C., and A.N. are named inventors on patent applications covering HCV-vectored vaccines and chimpanzee adenovirus vectors [WO 2006133911 (A3) hepatitis C virus nucleic acid vaccine, WO 2005071093 (A3) chimpanzee adenovirus vaccine carriers, WO 03031588 (A2) hepatitis C virus vaccine]. V.V. is an employee of GSK group of companies. The rest of the authors declare that there are no competing interests.
Figures

References
Grants and funding
- MRF-044-0001-RG-SWADL/MRF_/MRF_/United Kingdom
- MRF-044-0001-RG-SWADL/RCUK | MRC | Medical Research Foundation
- G0701694/MRC_/Medical Research Council/United Kingdom
- 109965/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- 2U19AI082630-06/U.S. Department of Health & Human Services | NIH | Center for Scientific Review (NIH Center for Scientific Review)
LinkOut - more resources
Full Text Sources
