

DNA Methylation at Birth Predicts Intellectual Functioning and Autism Features in Children with Fragile X Syndrome
- PMID: 33086711
- PMCID: PMC7589848
- DOI: 10.3390/ijms21207735
DNA Methylation at Birth Predicts Intellectual Functioning and Autism Features in Children with Fragile X Syndrome
Abstract
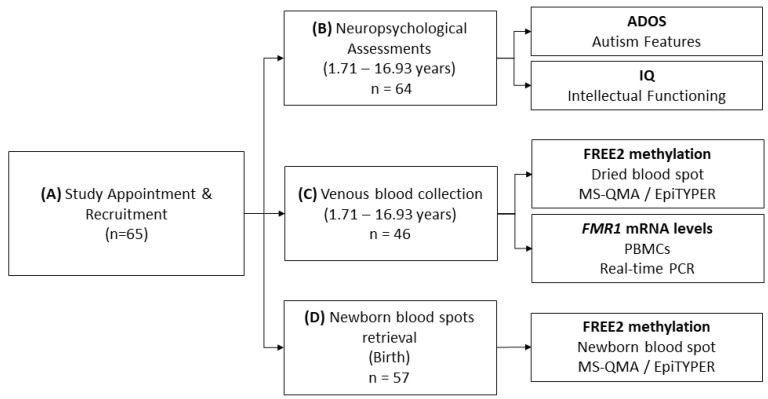
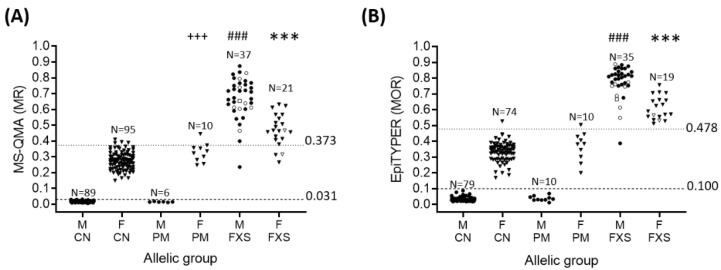
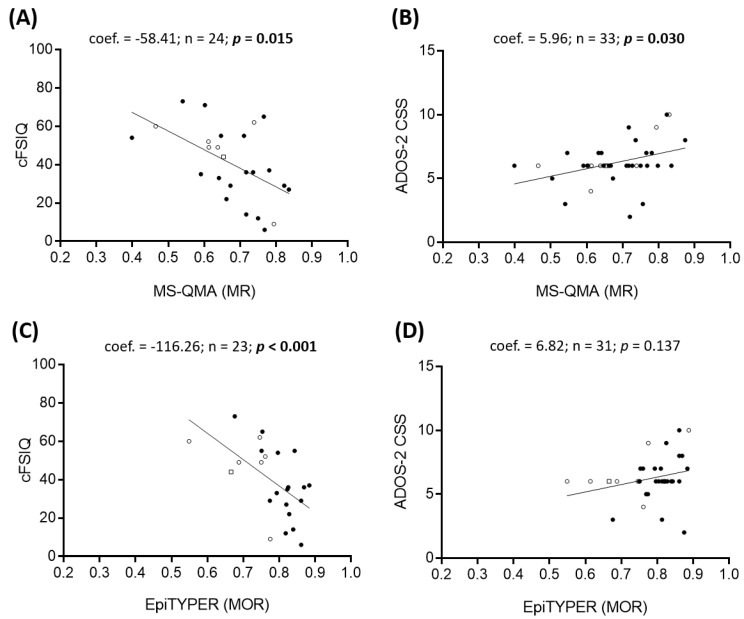
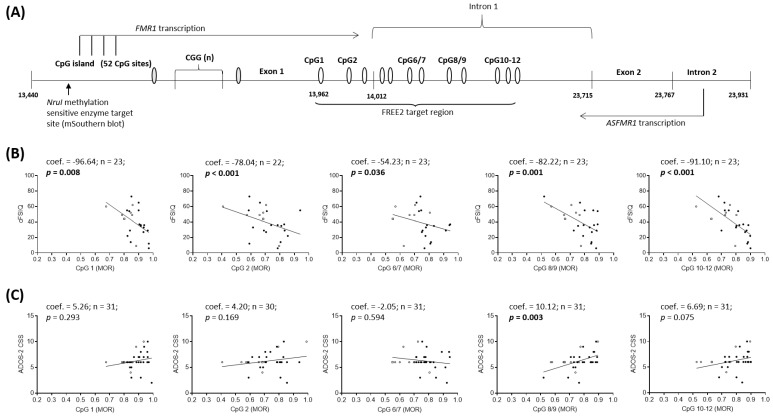
Fragile X syndrome (FXS) is a leading single-gene cause of intellectual disability (ID) with autism features. This study analysed diagnostic and prognostic utility of the Fragile X-Related Epigenetic Element 2 DNA methylation (FREE2m) assessed by Methylation Specific-Quantitative Melt Analysis and the EpiTYPER system, in retrospectively retrieved newborn blood spots (NBS) and newly created dried blood spots (DBS) from 65 children with FXS (~2-17 years). A further 168 NBS from infants from the general population were used to establish control reference ranges, in both sexes. FREE2m analysis showed sensitivity and specificity approaching 100%. In FXS males, NBS FREE2m strongly correlated with intellectual functioning and autism features, however associations were not as strong for FXS females. Fragile X mental retardation 1 gene (FMR1) mRNA levels in blood were correlated with FREE2m in both NBS and DBS, for both sexes. In females, DNAm was significantly increased at birth with a decrease in childhood. The findings support the use of FREE2m analysis in newborns for screening, diagnostic and prognostic testing in FXS.
Keywords: DNA methylation (DNAm); autism spectrum disorder (ASD); fragile X mental retardation 1 gene (FMR1 gene); fragile X syndrome (FXS); intellectual disability (ID); newborn screening.
Conflict of interest statement
David E. Godler is an inventor of the following patents: PCT/AU2010/001134; filing No. AU2010/903595; filing No. AU2011/902500; and filing No. 2013/900227, related to the technology described in this publication. David Eugeny Godler is the director of E.D.G. Innovations and Consulting Pty Ltd., Melbourne, Australia, that owns this intellectual property. The other authors declare no competing interests.
Figures
Similar articles
-
Incomplete silencing of full mutation alleles in males with fragile X syndrome is associated with autistic features.Mol Autism. 2019 May 3;10:21. doi: 10.1186/s13229-019-0271-7. eCollection 2019. Mol Autism. 2019. PMID: 31073396 Free PMC article.
-
Intellectual functioning and behavioural features associated with mosaicism in fragile X syndrome.J Neurodev Disord. 2019 Dec 26;11(1):41. doi: 10.1186/s11689-019-9288-7. J Neurodev Disord. 2019. PMID: 31878865 Free PMC article.
-
Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome.PLoS One. 2011;6(10):e26203. doi: 10.1371/journal.pone.0026203. Epub 2011 Oct 12. PLoS One. 2011. PMID: 22022567 Free PMC article.
-
Lessons from fragile X regarding neurobiology, autism, and neurodegeneration.J Dev Behav Pediatr. 2006 Feb;27(1):63-74. doi: 10.1097/00004703-200602000-00012. J Dev Behav Pediatr. 2006. PMID: 16511373 Review.
-
DNA Methylation, Mechanisms of FMR1 Inactivation and Therapeutic Perspectives for Fragile X Syndrome.Biomolecules. 2021 Feb 16;11(2):296. doi: 10.3390/biom11020296. Biomolecules. 2021. PMID: 33669384 Free PMC article. Review.
Cited by
-
Fragile X Mental Retardation Protein and Cerebral Expression of Metabotropic Glutamate Receptor Subtype 5 in Men with Fragile X Syndrome: A Pilot Study.Brain Sci. 2022 Feb 26;12(3):314. doi: 10.3390/brainsci12030314. Brain Sci. 2022. PMID: 35326270 Free PMC article.
-
High-throughput assessment of FMR1 and SNRPN methylation-based newborn screening using IsoPure and QIAcube HT systems.Epigenomics. 2025 Sep;17(13):851-863. doi: 10.1080/17501911.2025.2544530. Epub 2025 Aug 13. Epigenomics. 2025. PMID: 40801290 Free PMC article.
-
Tissue mosaicism, FMR1 expression and intellectual functioning in males with fragile X syndrome.Am J Med Genet A. 2023 Feb;191(2):357-369. doi: 10.1002/ajmg.a.63027. Epub 2022 Nov 8. Am J Med Genet A. 2023. PMID: 36349505 Free PMC article.
-
Parental Reports on Early Autism Behaviors in Their Children with Fragile X Syndrome as a Function of Infant Feeding.Nutrients. 2021 Aug 22;13(8):2888. doi: 10.3390/nu13082888. Nutrients. 2021. PMID: 34445048 Free PMC article.
-
A near normal distribution of IQ in Fragile X Syndrome.Sci Rep. 2024 Oct 4;14(1):23058. doi: 10.1038/s41598-024-73626-y. Sci Rep. 2024. PMID: 39367109 Free PMC article.
References
-
- Joubert B.R., Felix J.F., Yousefi P., Bakulski K.M., Just A.C., Breton C., Reese S.E., Markunas C.A., Richmond R.C., Xu C.-J., et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016;98:680–696. doi: 10.1016/j.ajhg.2016.02.019. - DOI - PMC - PubMed
-
- Aref-Eshghi E., Rodenhiser D.I., Schenkel L.C., Lin H., Skinner C., Ainsworth P., Paré G., Hood R.L., Bulman D.E., Kernohan K.D., et al. Genomic DNA Methylation Signatures Enable Concurrent Diagnosis and Clinical Genetic Variant Classification in Neurodevelopmental Syndromes. Am. J. Hum. Genet. 2018;102:156–174. doi: 10.1016/j.ajhg.2017.12.008. - DOI - PMC - PubMed
-
- Andrews S.V., Ellis S.E., Bakulski K.M., Sheppard B., Croen L.A., Hertz-Picciotto I., Newschaffer C.J., Feinberg A.P., Arking D.E., Ladd-Acosta C., et al. Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder. Nat. Commun. 2017;8:1011. doi: 10.1038/s41467-017-00868-y. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
