

Selection of Oncogenic Mutant Clones in Normal Human Skin Varies with Body Site
- PMID: 33087317
- PMCID: PMC7116717
- DOI: 10.1158/2159-8290.CD-20-1092
Selection of Oncogenic Mutant Clones in Normal Human Skin Varies with Body Site
Abstract
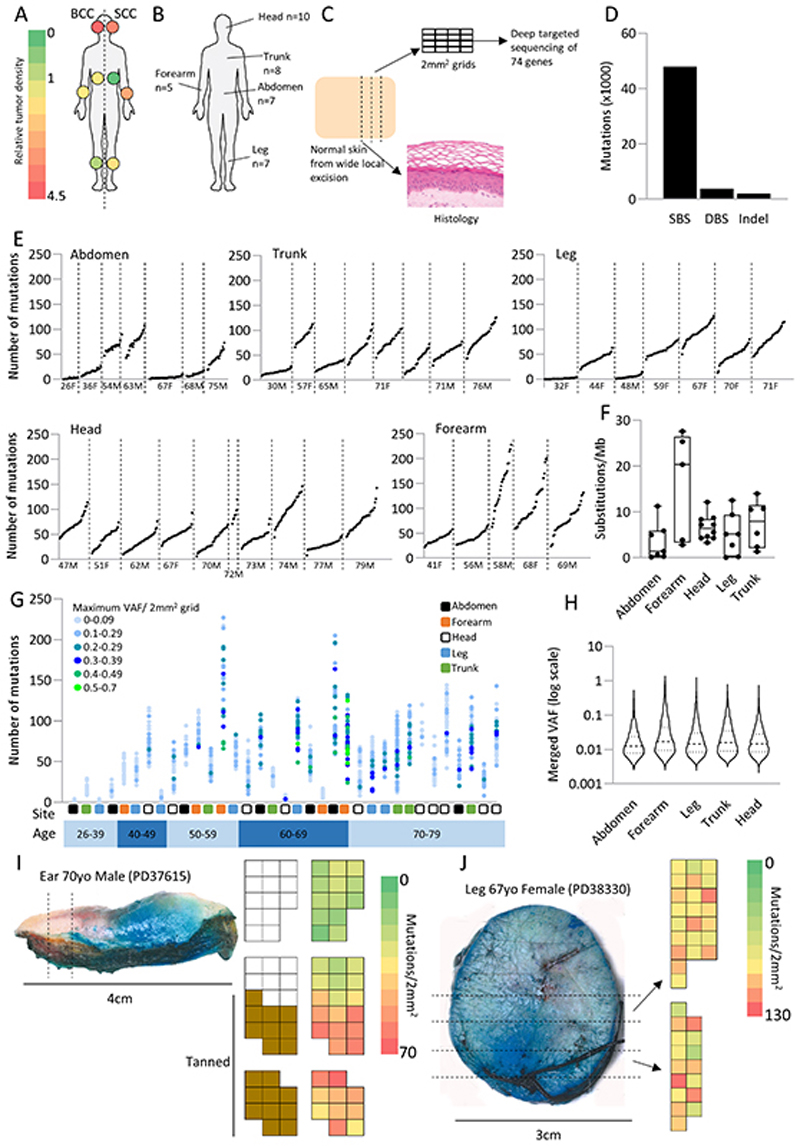
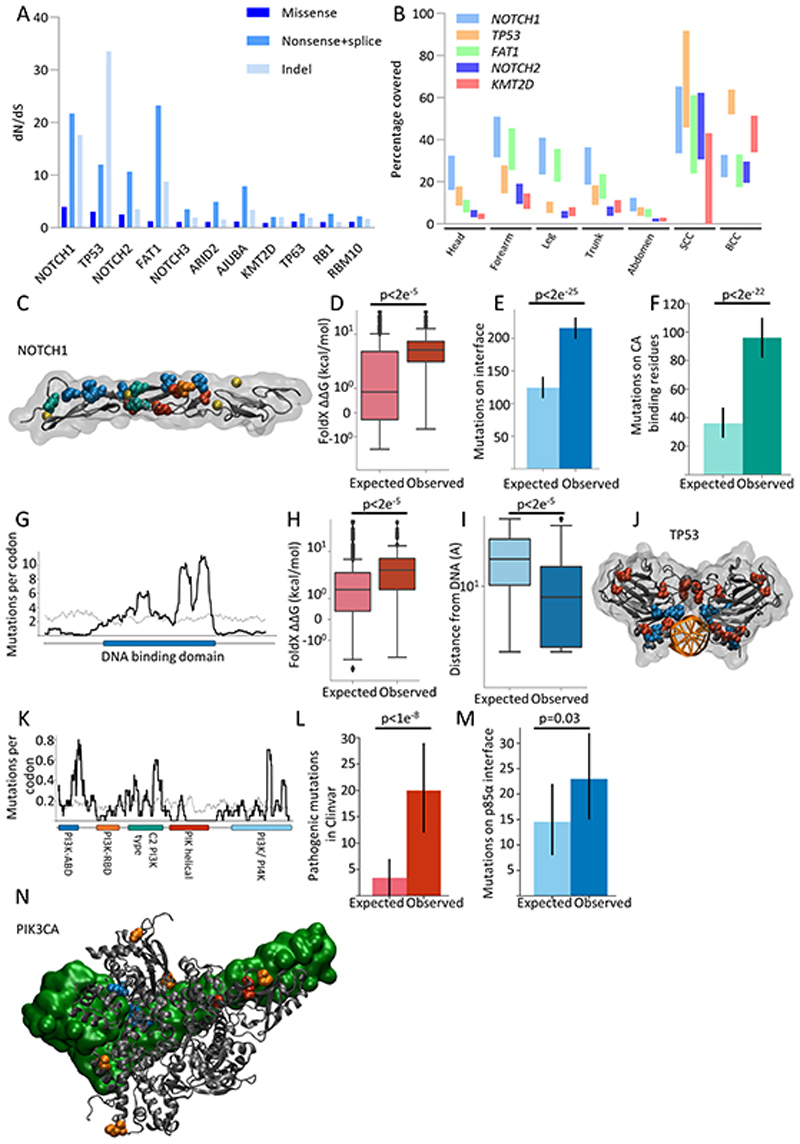
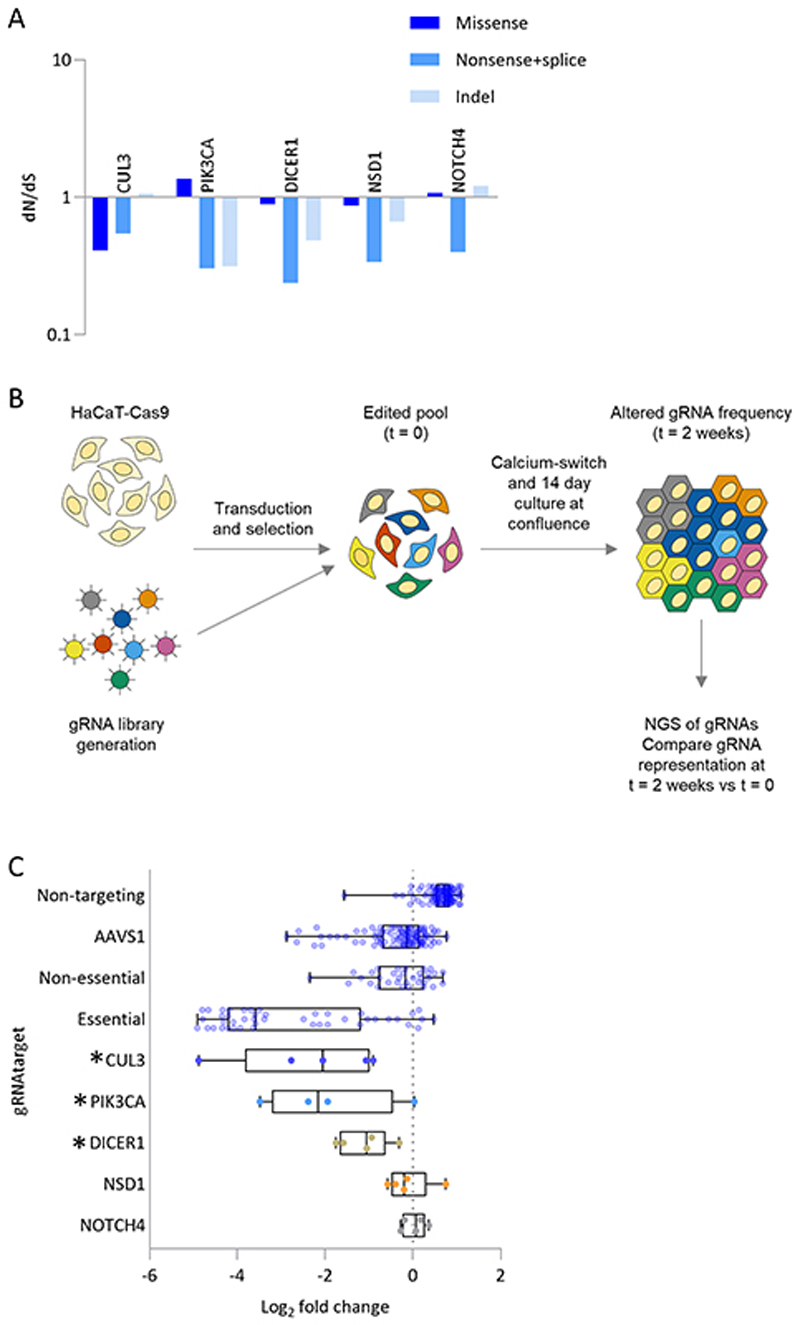
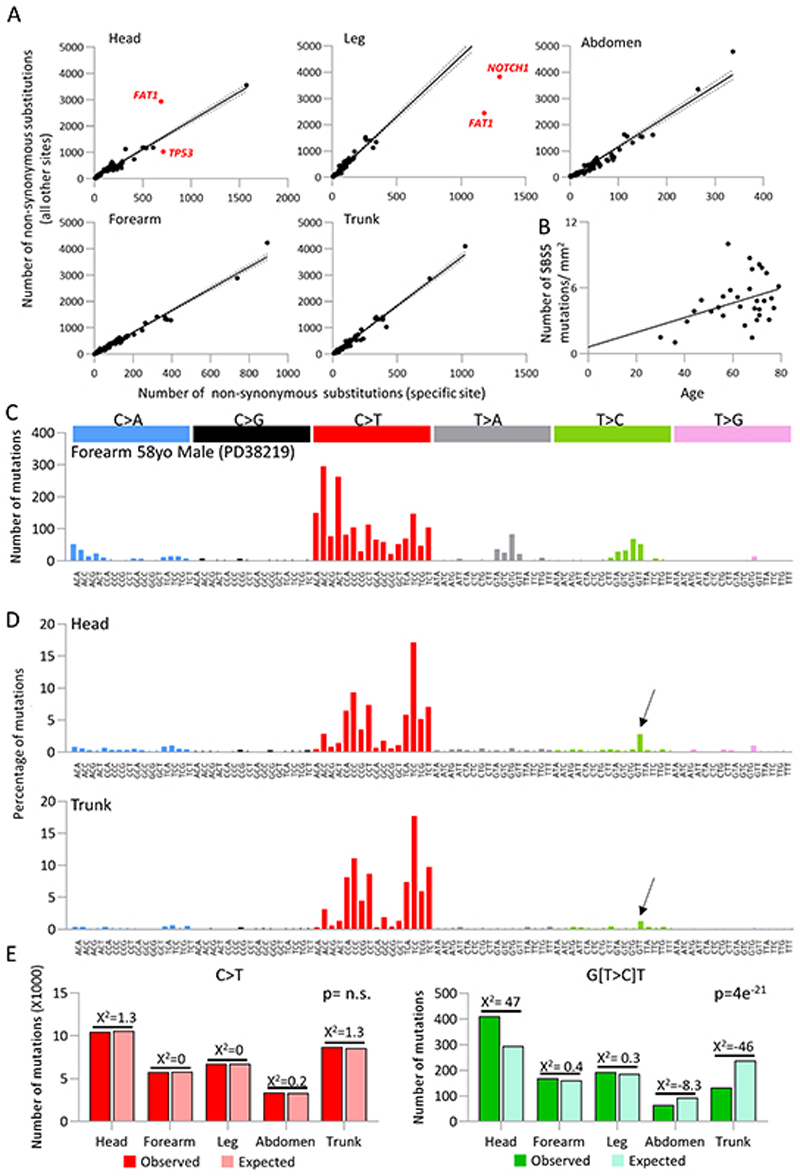
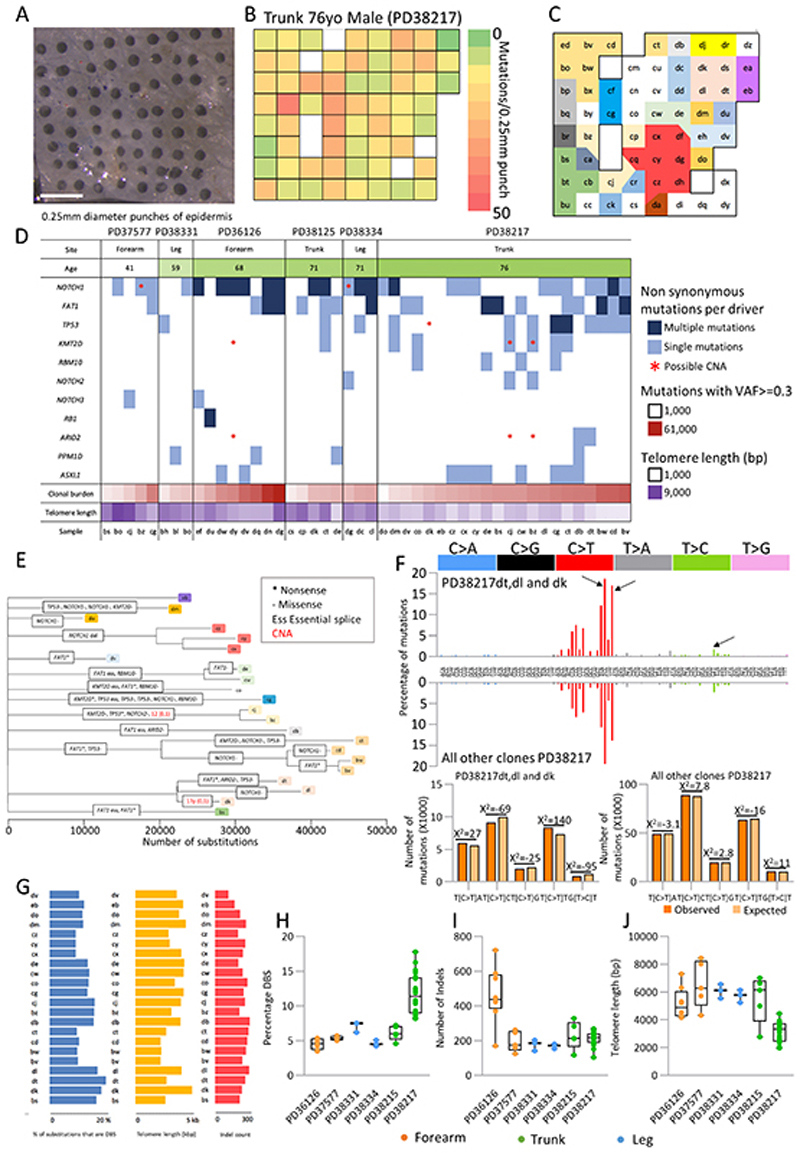
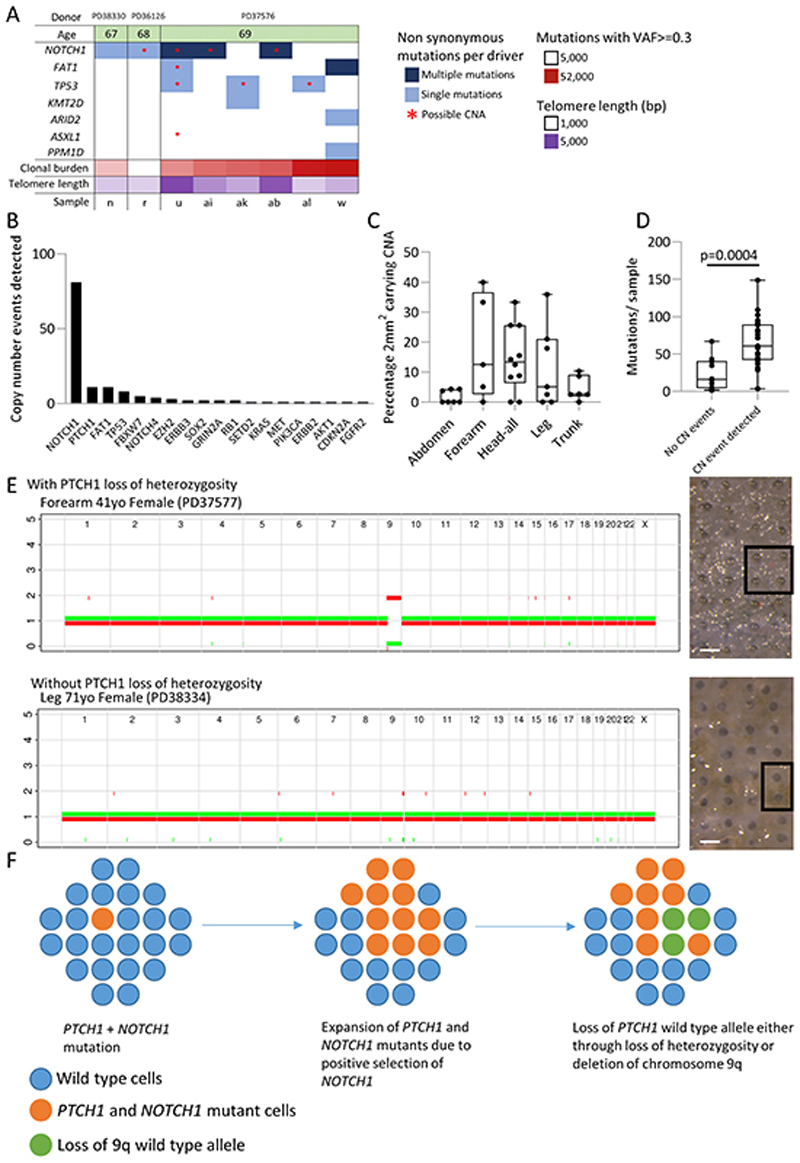
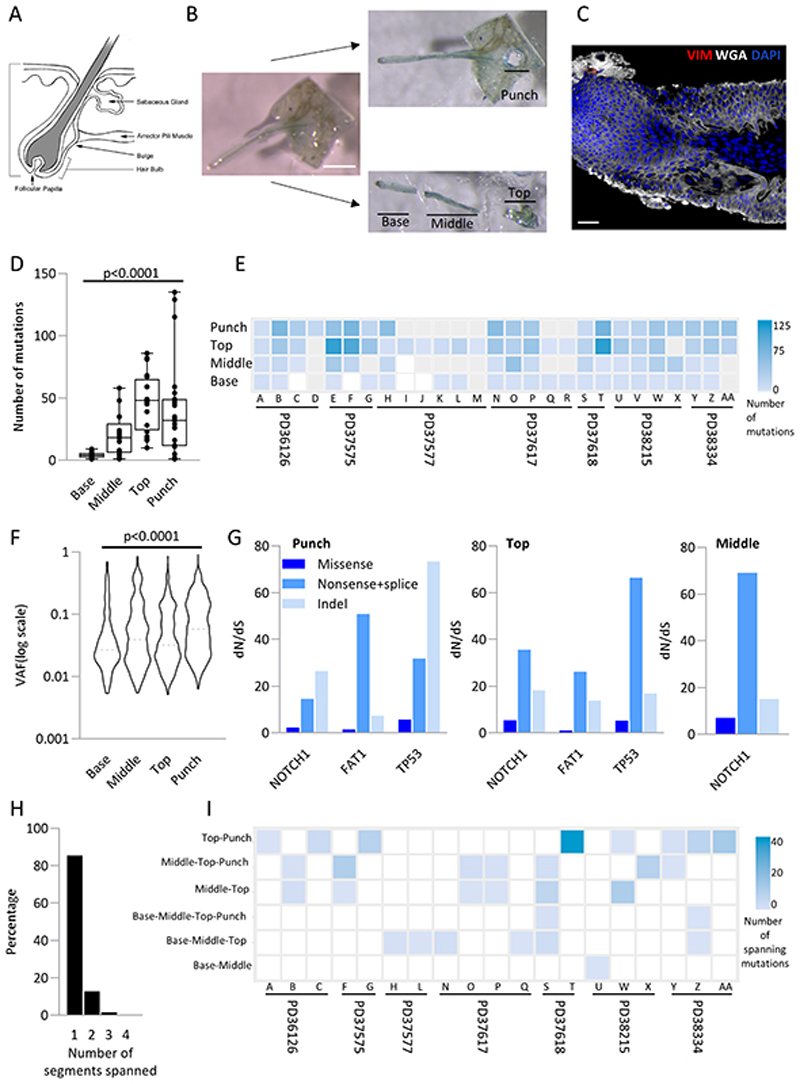
Skin cancer risk varies substantially across the body, yet how this relates to the mutations found in normal skin is unknown. Here we mapped mutant clones in skin from high- and low-risk sites. The density of mutations varied by location. The prevalence of NOTCH1 and FAT1 mutations in forearm, trunk, and leg skin was similar to that in keratinocyte cancers. Most mutations were caused by ultraviolet light, but mutational signature analysis suggested differences in DNA-repair processes between sites. Eleven mutant genes were under positive selection, with TP53 preferentially selected in the head and FAT1 in the leg. Fine-scale mapping revealed 10% of clones had copy-number alterations. Analysis of hair follicles showed mutations in the upper follicle resembled adjacent skin, but the lower follicle was sparsely mutated. Normal skin is a dense patchwork of mutant clones arising from competitive selection that varies by location. SIGNIFICANCE: Mapping mutant clones across the body reveals normal skin is a dense patchwork of mutant cells. The variation in cancer risk between sites substantially exceeds that in mutant clone density. More generally, mutant genes cannot be assigned as cancer drivers until their prevalence in normal tissue is known.See related commentary by De Dominici and DeGregori, p. 227.This article is highlighted in the In This Issue feature, p. 211.
©2020 American Association for Cancer Research.
Conflict of interest statement
The authors declare no competing interests.
Conflict of Interest: The authors declare no potential conflicts of interest.
Figures
Comment in
-
Shedding Light on Mutant Clonal Dynamics and Cancer Risk in the Skin.Cancer Discov. 2021 Feb;11(2):227-229. doi: 10.1158/2159-8290.CD-20-1644. Cancer Discov. 2021. PMID: 33531425
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
