

Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics
- PMID: 33087716
- PMCID: PMC7577981
- DOI: 10.1038/s41467-020-19119-8
Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics
Erratum in
-
Publisher Correction: Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics.Nat Commun. 2020 Nov 19;11(1):5996. doi: 10.1038/s41467-020-19902-7. Nat Commun. 2020. PMID: 33214561 Free PMC article.
-
Author Correction: Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics.Nat Commun. 2021 May 10;12(1):2823. doi: 10.1038/s41467-021-23280-z. Nat Commun. 2021. PMID: 33972555 Free PMC article. No abstract available.
Abstract
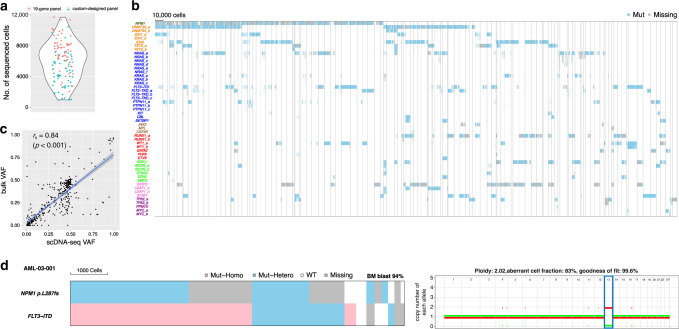
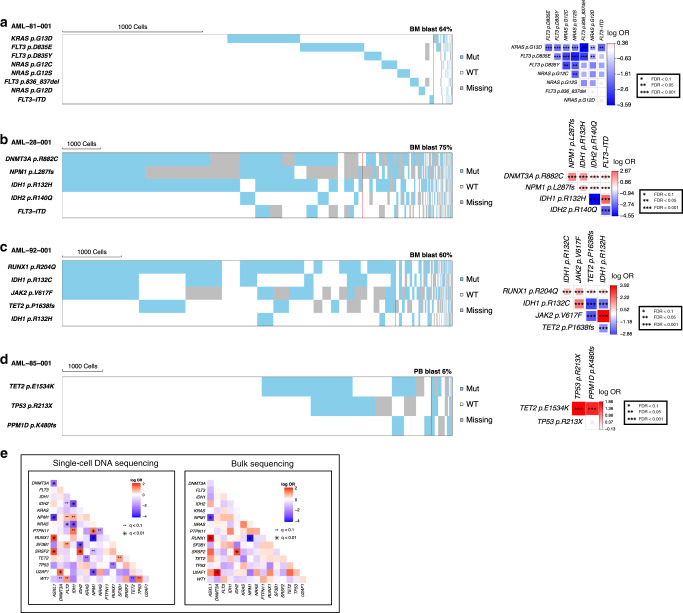
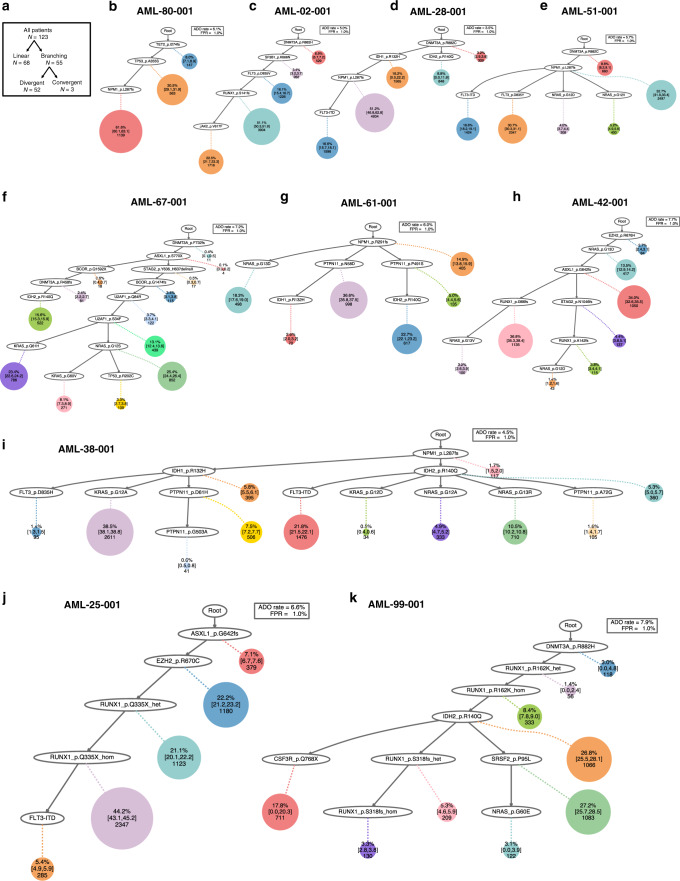
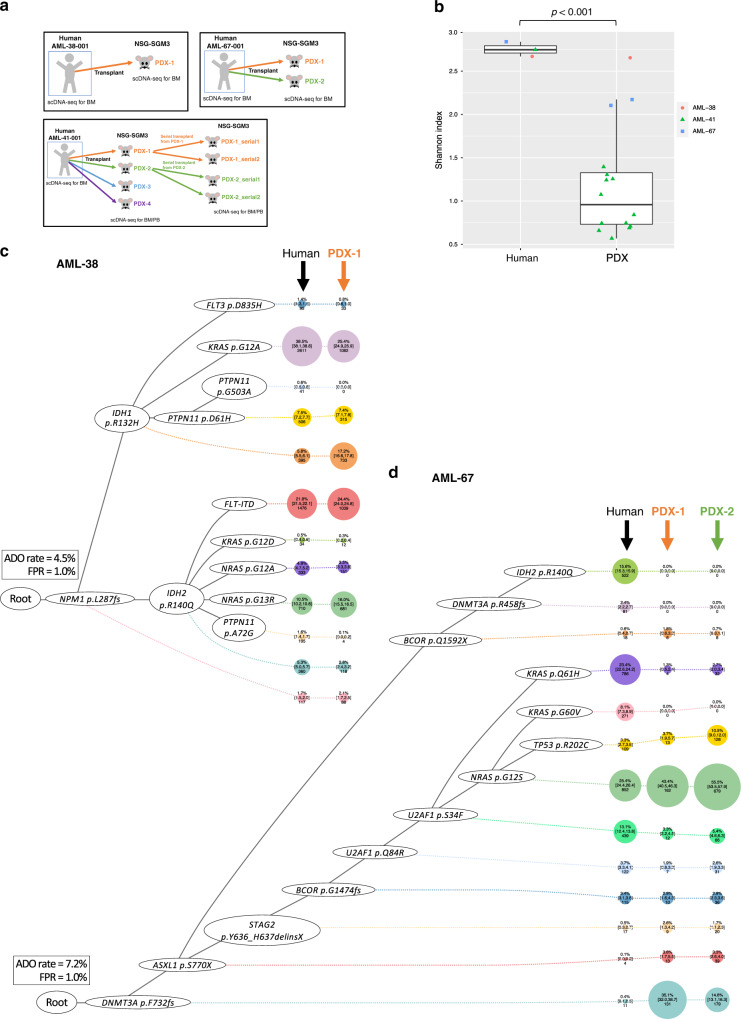
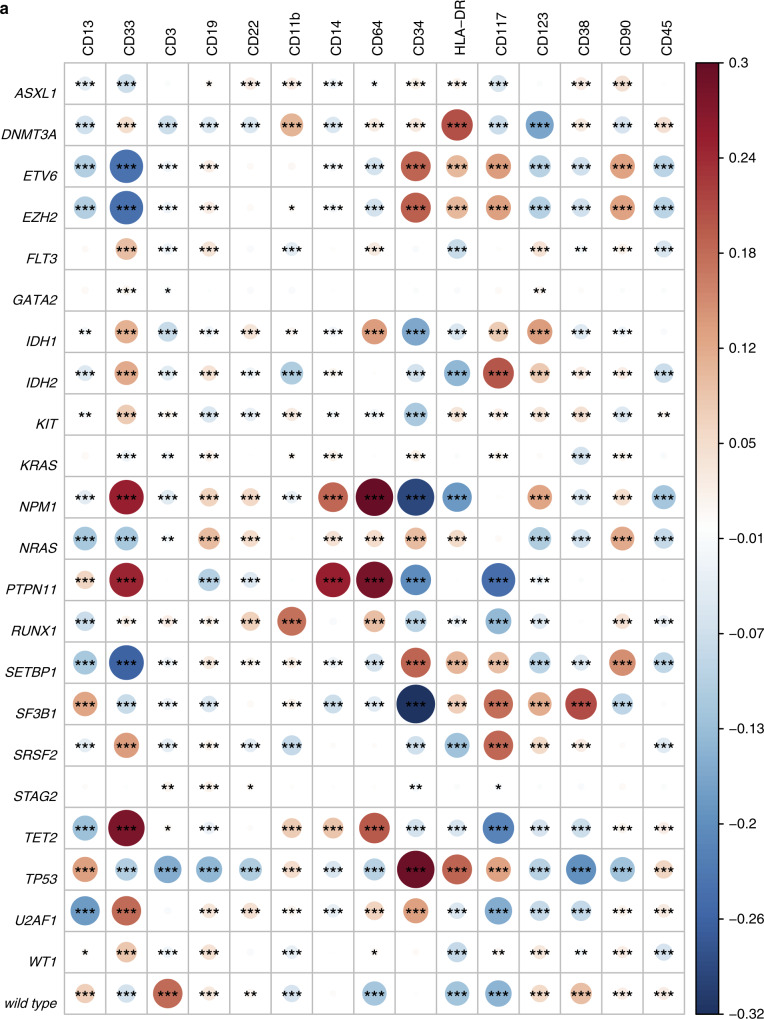
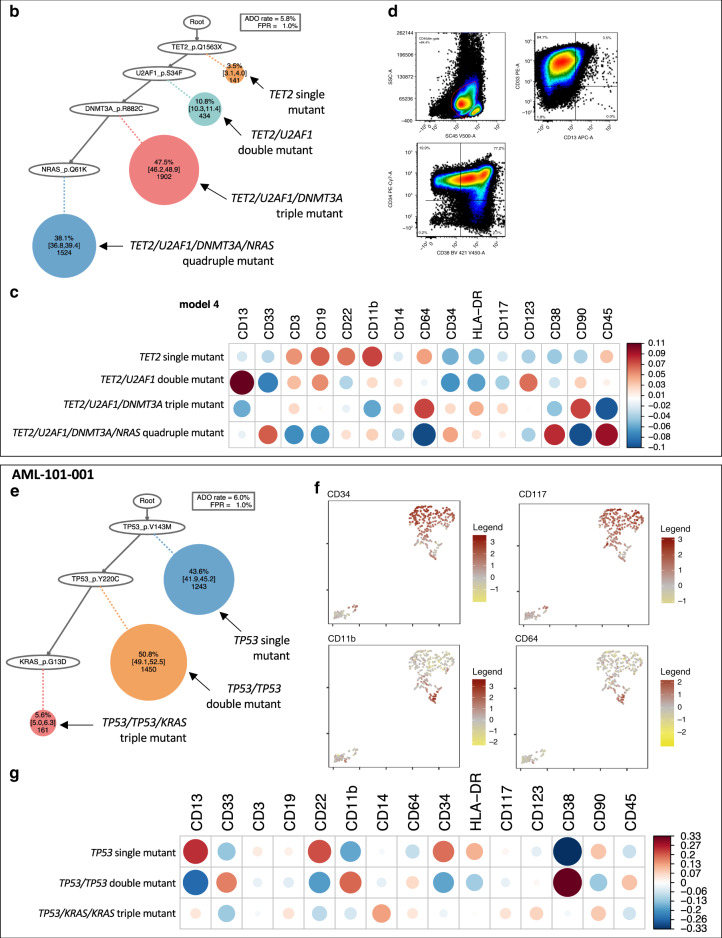
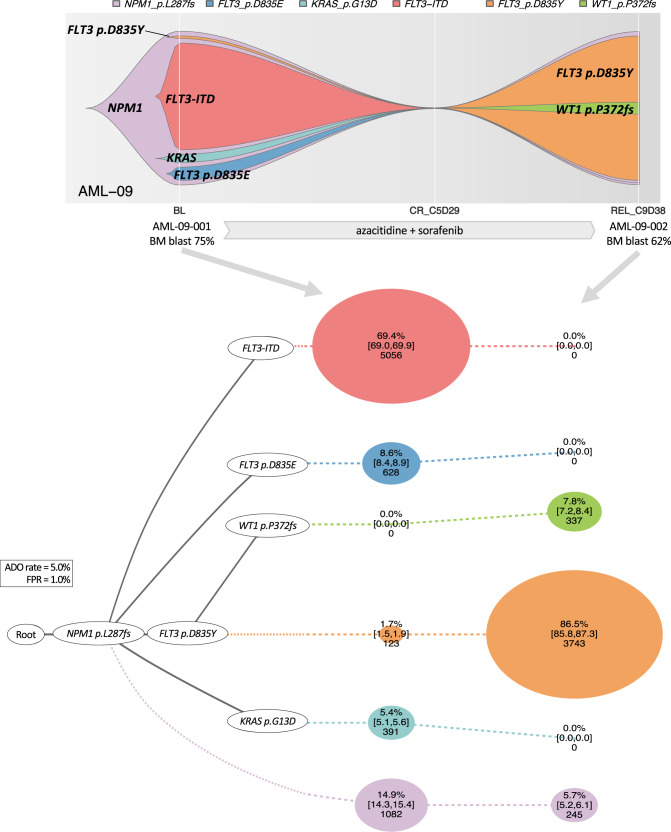
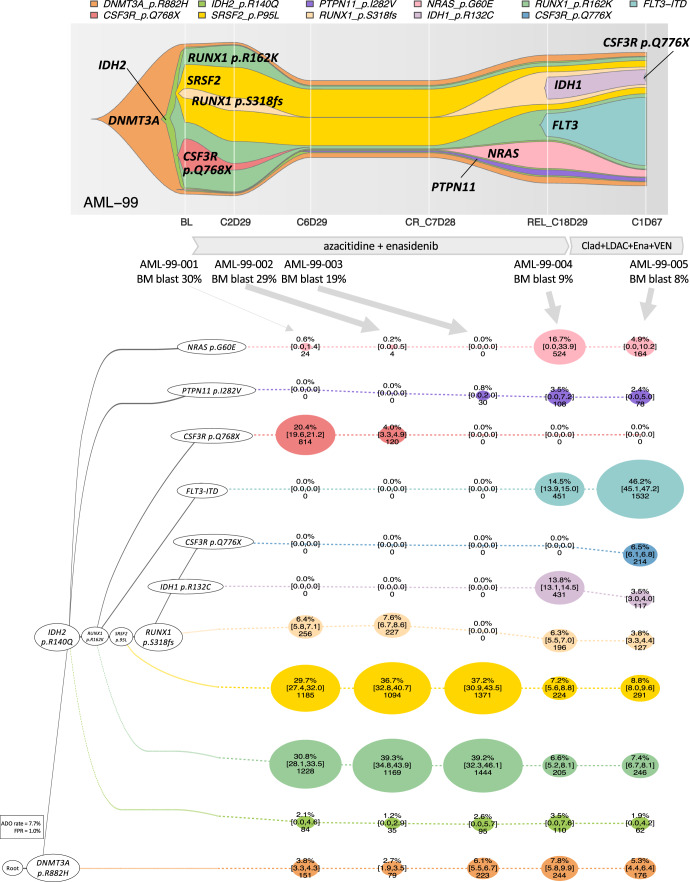
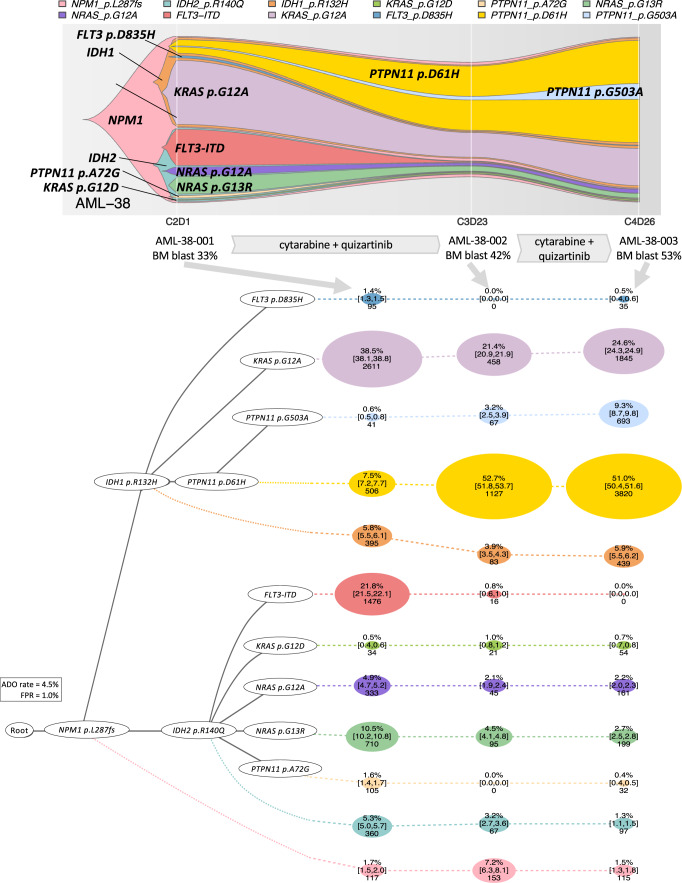
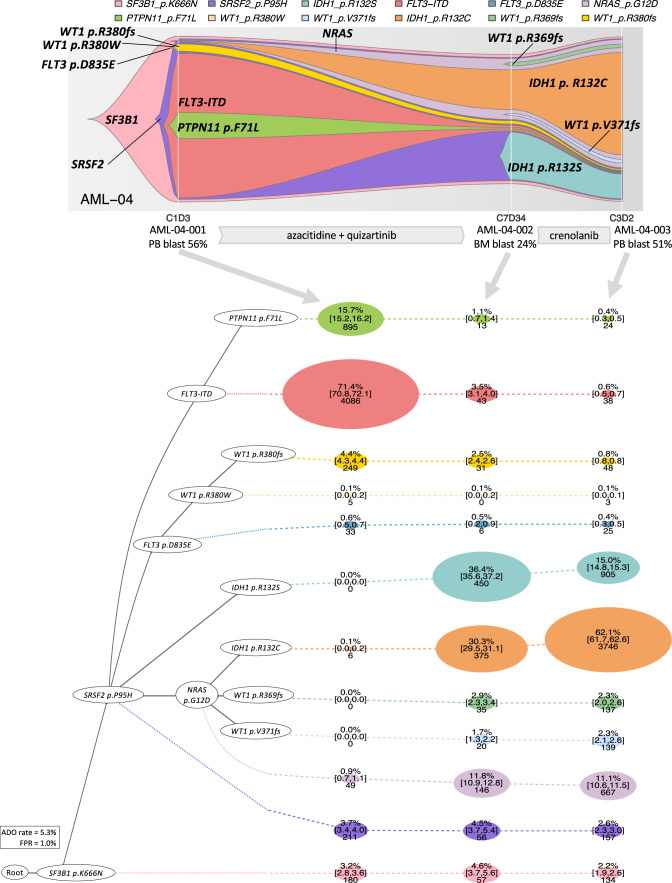
Clonal diversity is a consequence of cancer cell evolution driven by Darwinian selection. Precise characterization of clonal architecture is essential to understand the evolutionary history of tumor development and its association with treatment resistance. Here, using a single-cell DNA sequencing, we report the clonal architecture and mutational histories of 123 acute myeloid leukemia (AML) patients. The single-cell data reveals cell-level mutation co-occurrence and enables reconstruction of mutational histories characterized by linear and branching patterns of clonal evolution, with the latter including convergent evolution. Through xenotransplantion, we show leukemia initiating capabilities of individual subclones evolving in parallel. Also, by simultaneous single-cell DNA and cell surface protein analysis, we illustrate both genetic and phenotypic evolution in AML. Lastly, single-cell analysis of longitudinal samples reveals underlying evolutionary process of therapeutic resistance. Together, these data unravel clonal diversity and evolution patterns of AML, and highlight their clinical relevance in the era of precision medicine.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
