

Early-onset autoimmunity associated with SOCS1 haploinsufficiency
- PMID: 33087723
- PMCID: PMC7578789
- DOI: 10.1038/s41467-020-18925-4
Early-onset autoimmunity associated with SOCS1 haploinsufficiency
Abstract
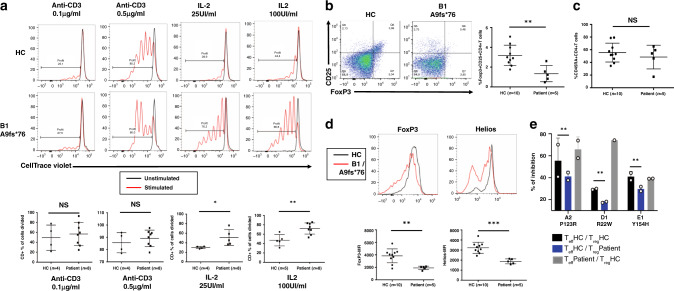
Autoimmunity can occur when a checkpoint of self-tolerance fails. The study of familial autoimmune diseases can reveal pathophysiological mechanisms involved in more common autoimmune diseases. Here, by whole-exome/genome sequencing we identify heterozygous, autosomal-dominant, germline loss-of-function mutations in the SOCS1 gene in ten patients from five unrelated families with early onset autoimmune manifestations. The intracellular protein SOCS1 is known to downregulate cytokine signaling by inhibiting the JAK-STAT pathway. Accordingly, patient-derived lymphocytes exhibit increased STAT activation in vitro in response to interferon-γ, IL-2 and IL-4 that is reverted by the JAK1/JAK2 inhibitor ruxolitinib. This effect is associated with a series of in vitro and in vivo immune abnormalities consistent with lymphocyte hyperactivity. Hence, SOCS1 haploinsufficiency causes a dominantly inherited predisposition to early onset autoimmune diseases related to cytokine hypersensitivity of immune cells.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
