

Immune receptor inhibition through enforced phosphatase recruitment
- PMID: 33087934
- PMCID: PMC7875542
- DOI: 10.1038/s41586-020-2851-2
Immune receptor inhibition through enforced phosphatase recruitment
Abstract
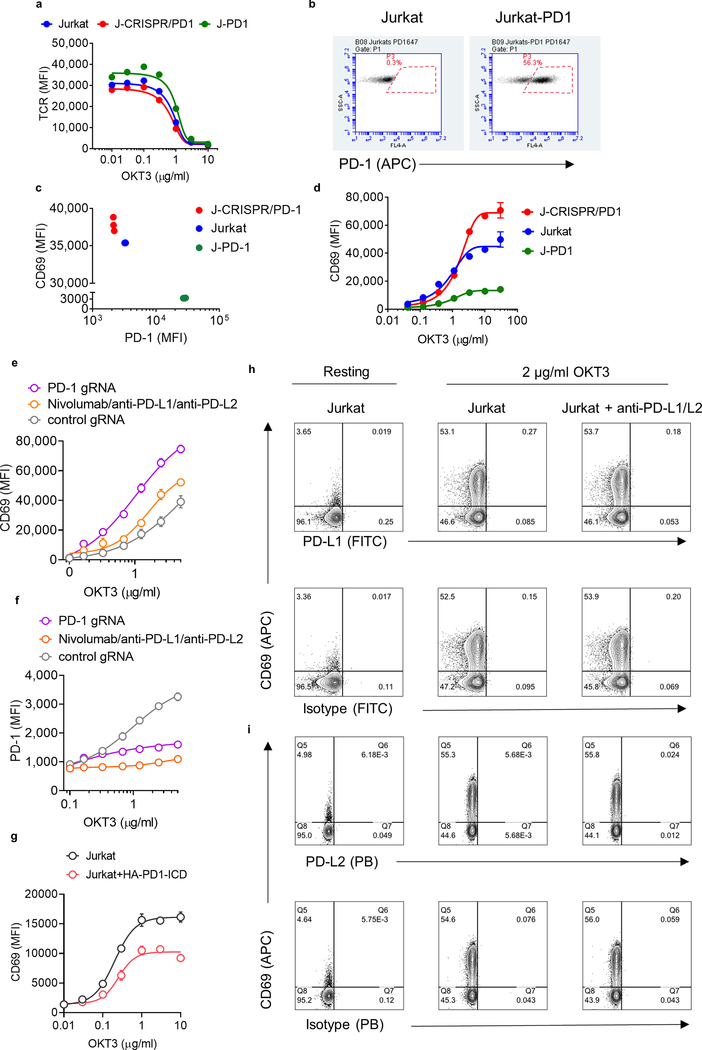
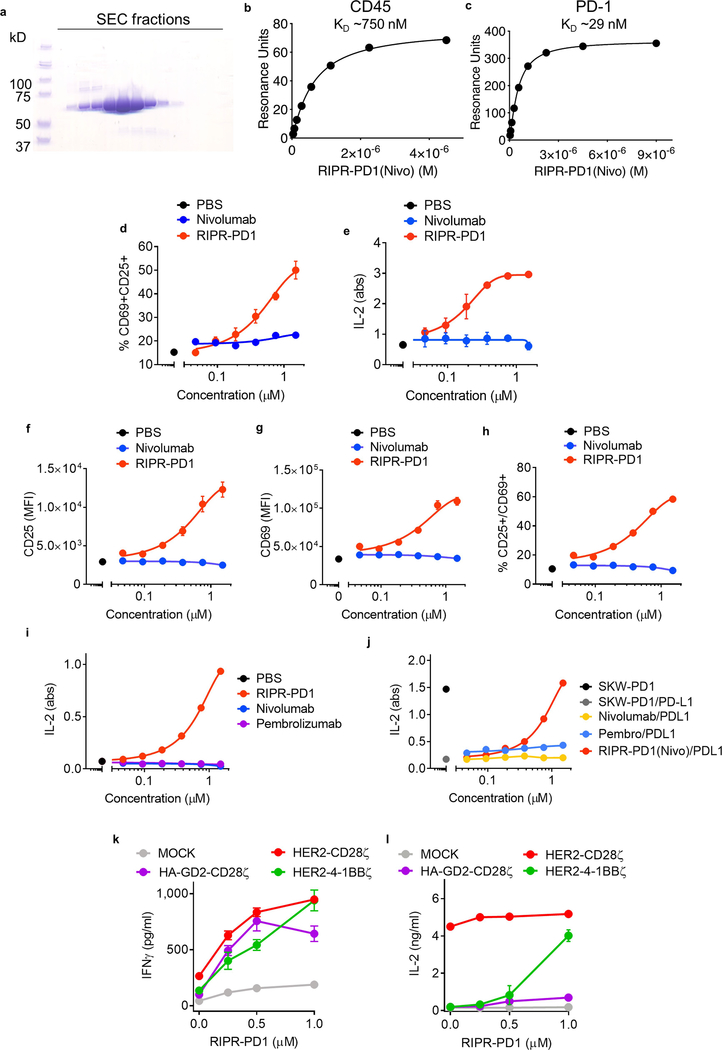
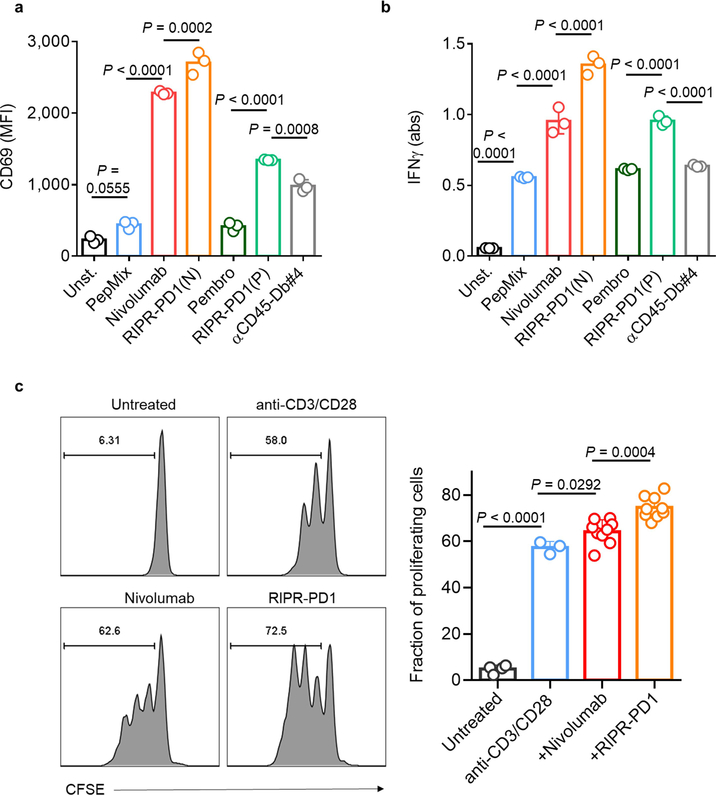
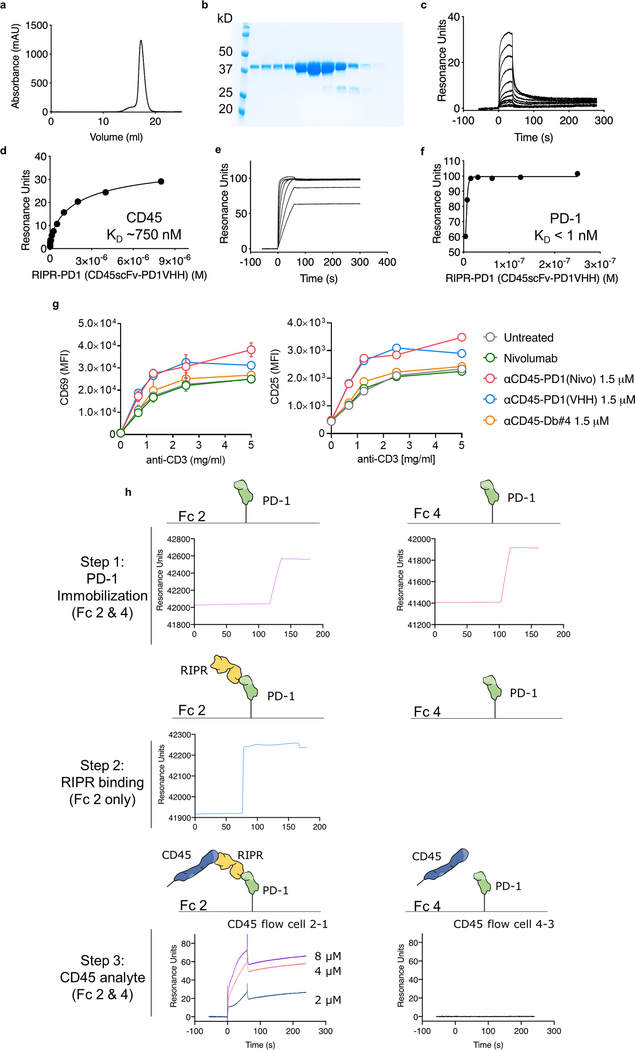
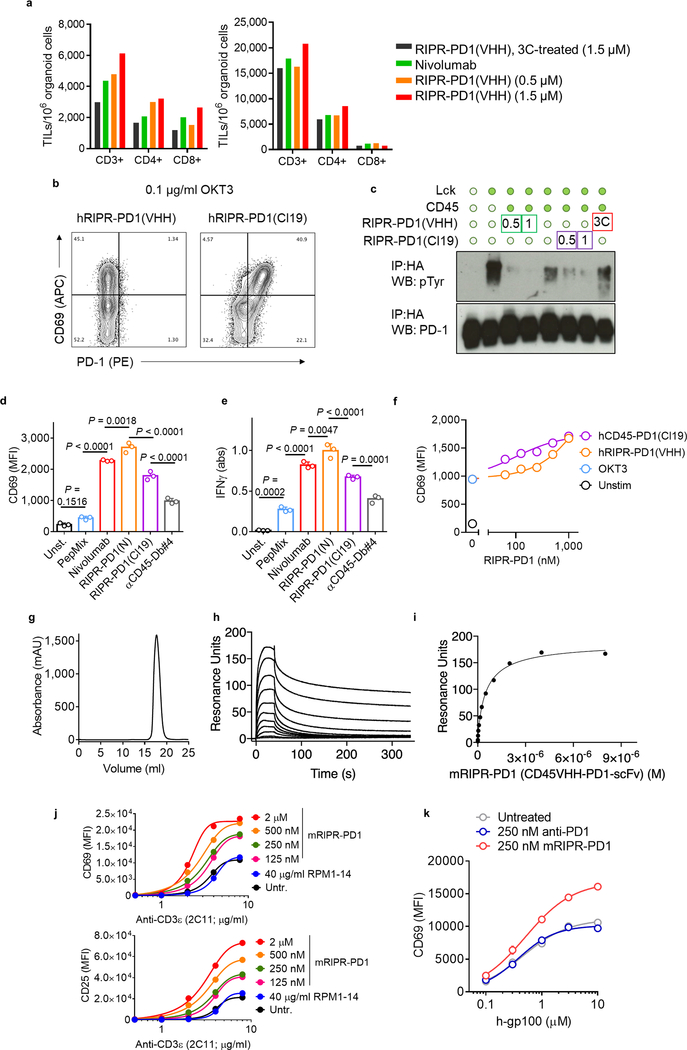
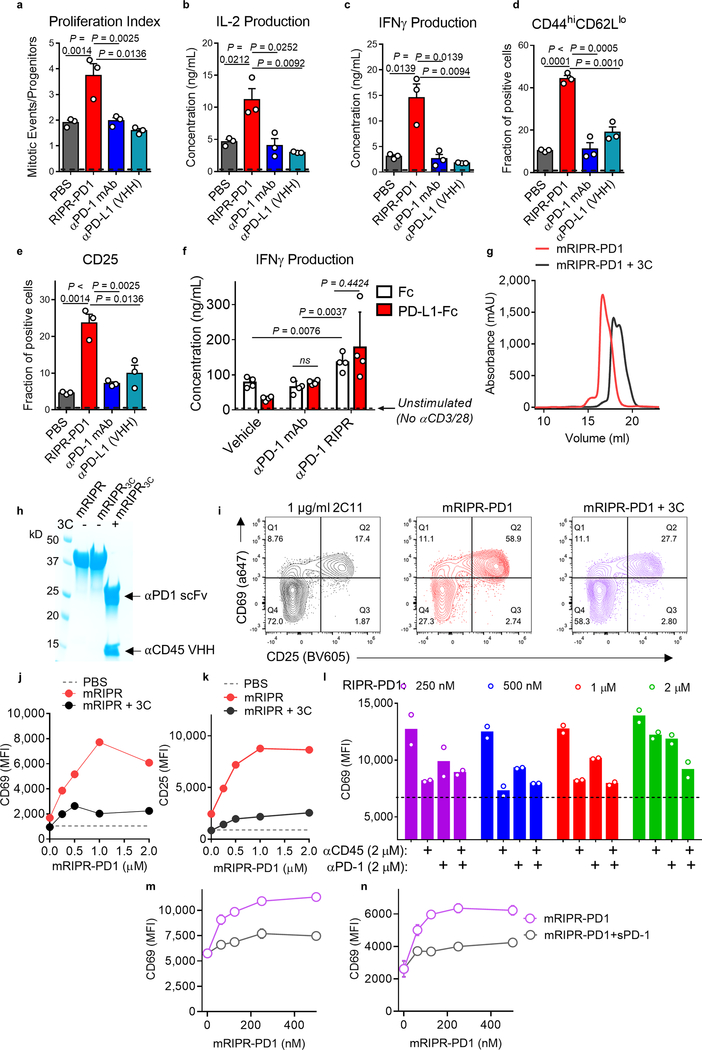
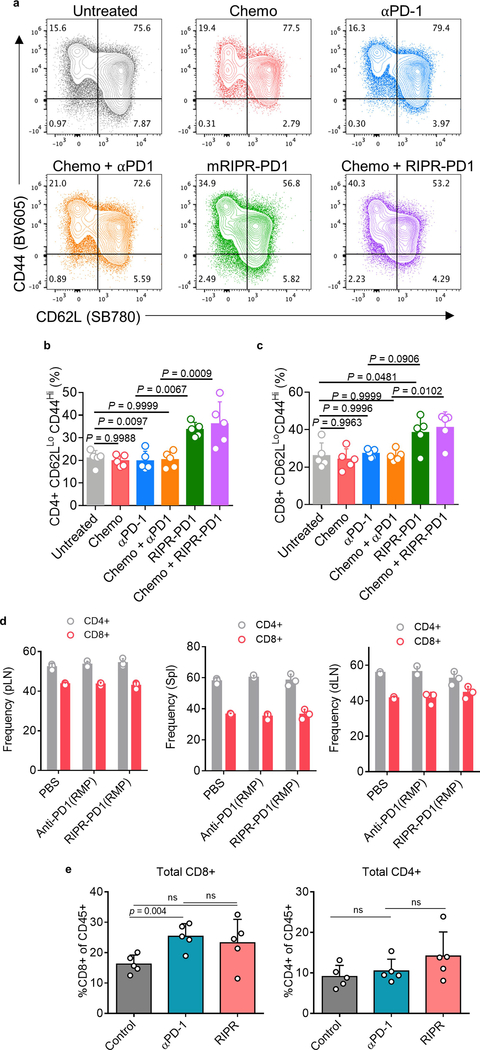
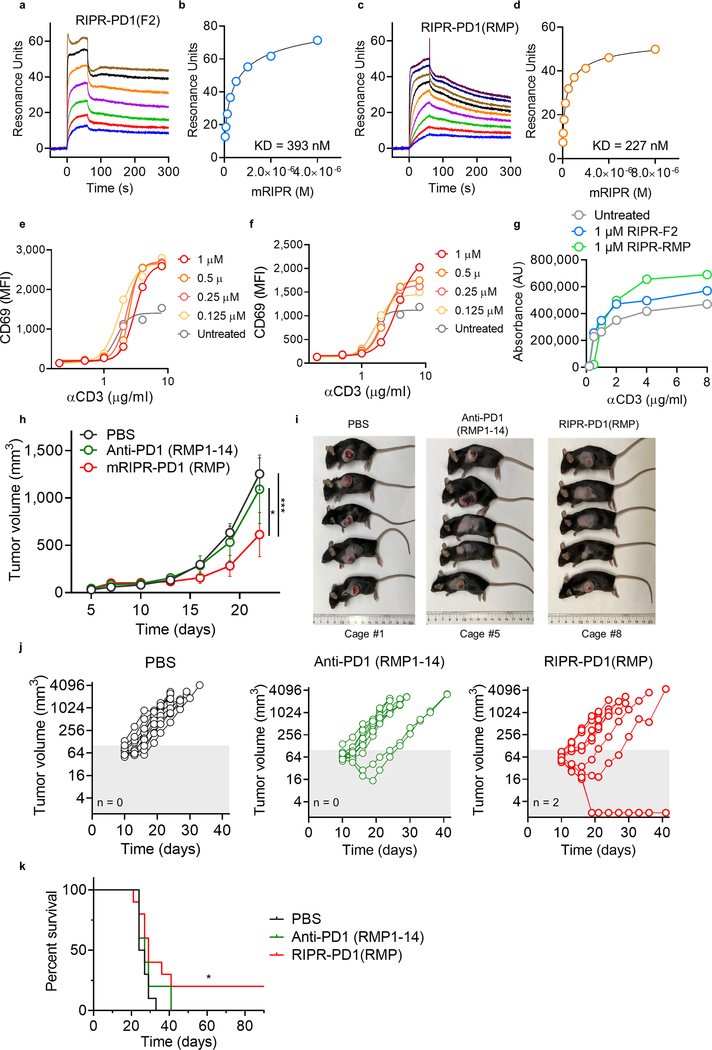
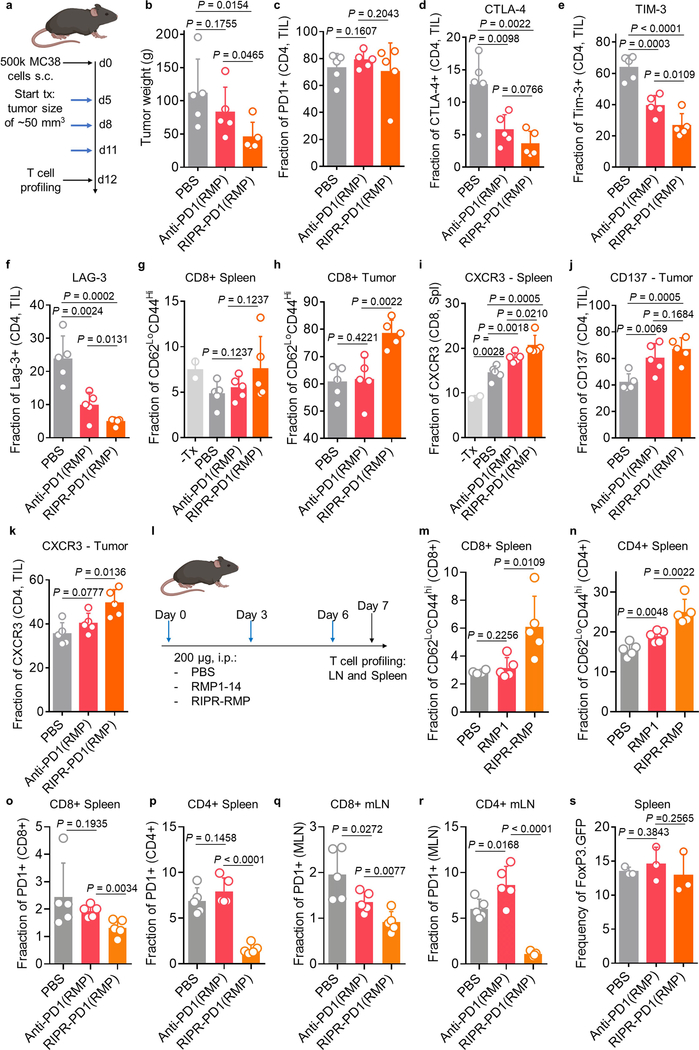
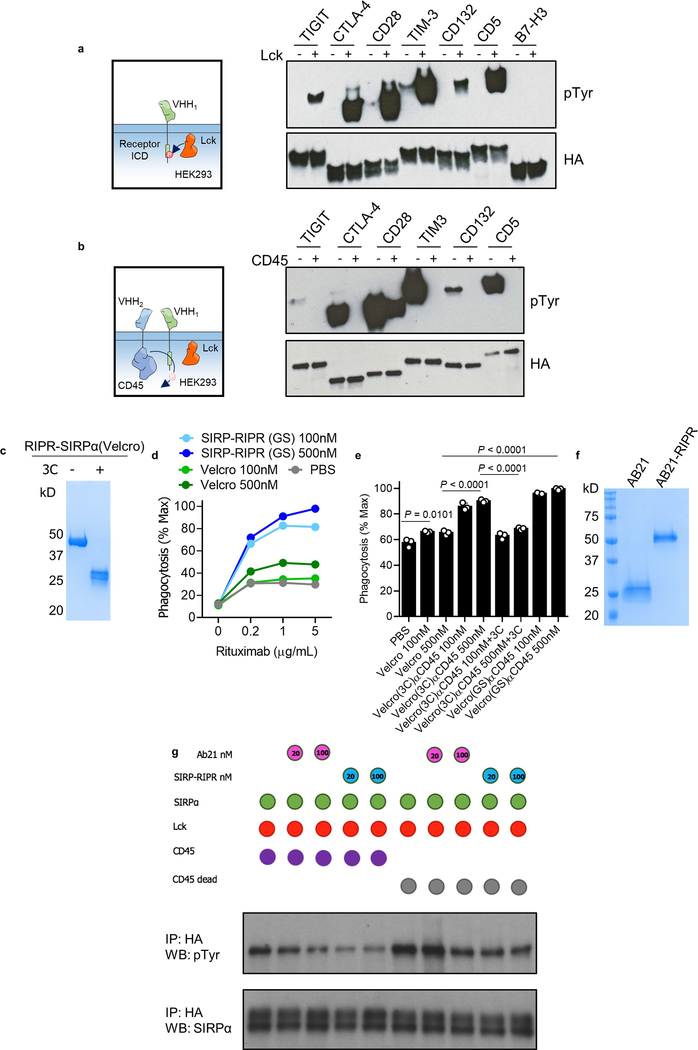
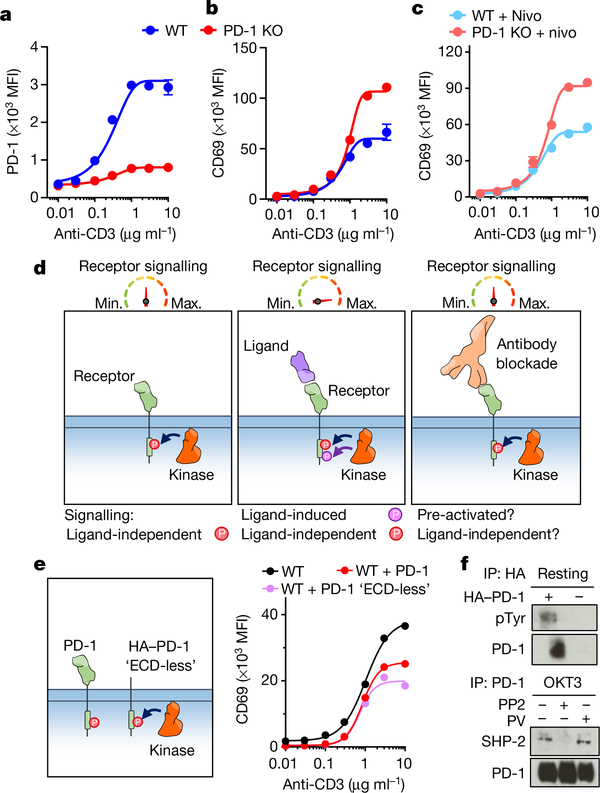
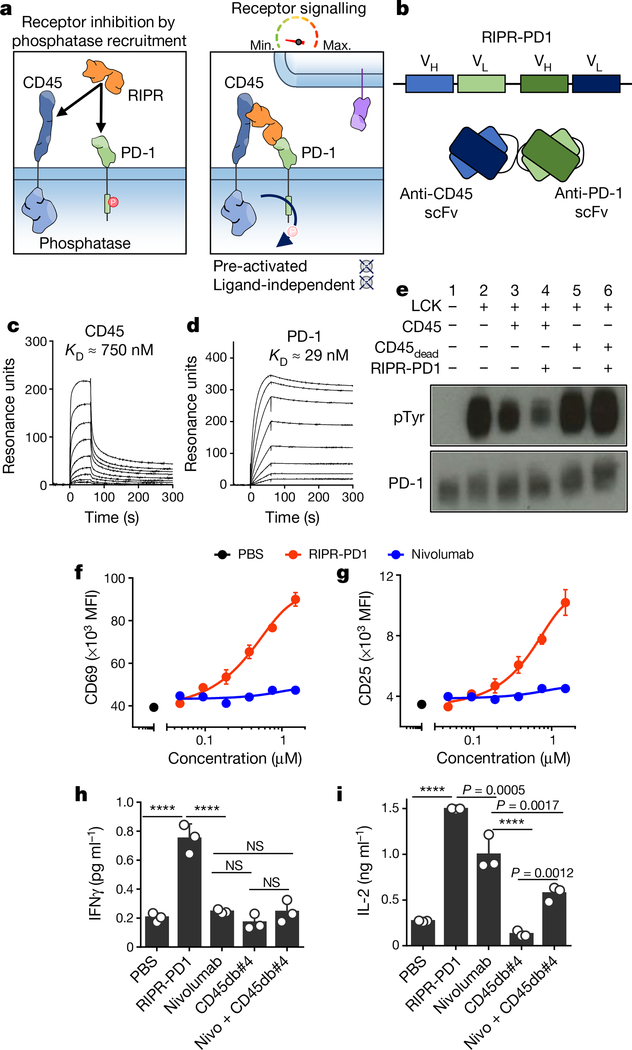
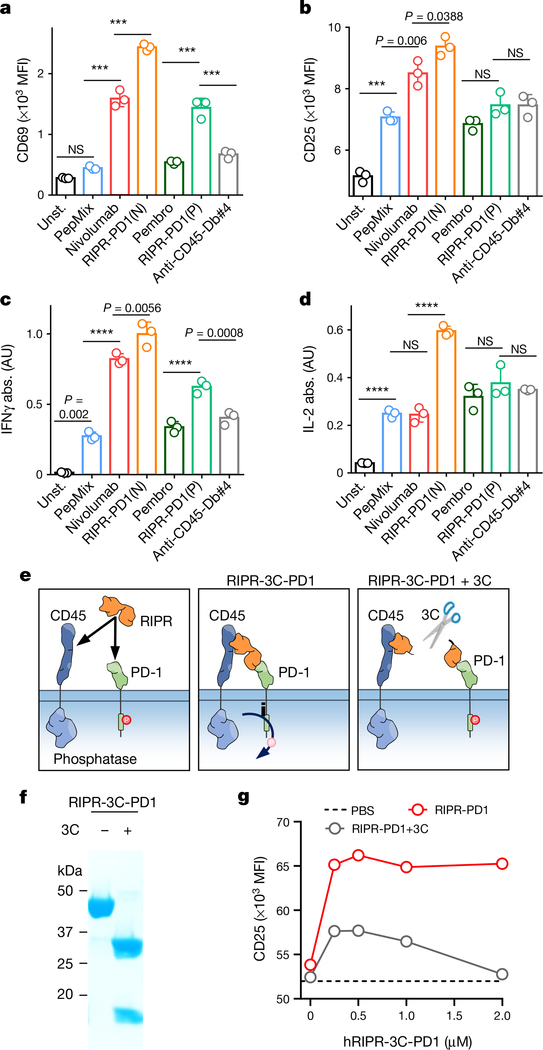
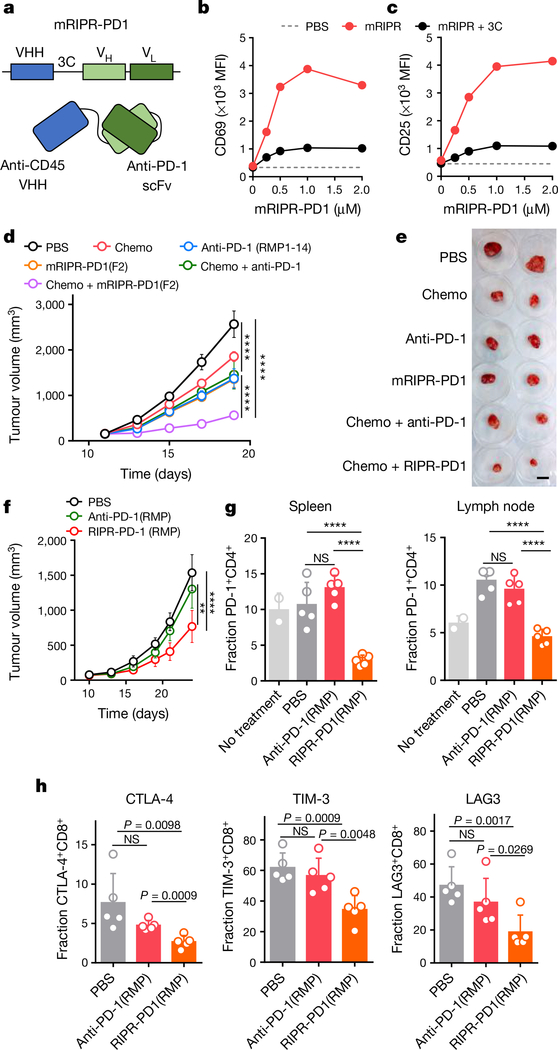
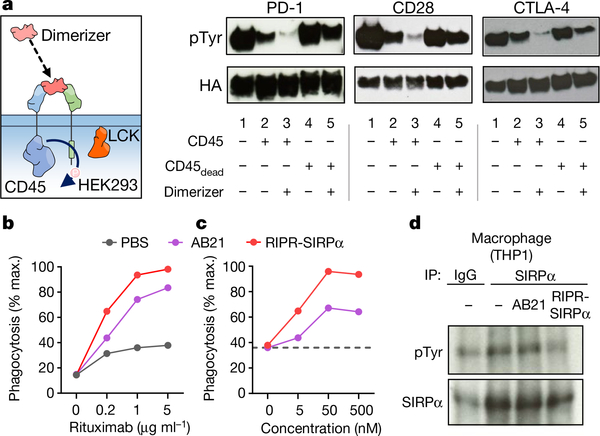
Antibodies that antagonize extracellular receptor-ligand interactions are used as therapeutic agents for many diseases to inhibit signalling by cell-surface receptors1. However, this approach does not directly prevent intracellular signalling, such as through tonic or sustained signalling after ligand engagement. Here we present an alternative approach for attenuating cell-surface receptor signalling, termed receptor inhibition by phosphatase recruitment (RIPR). This approach compels cis-ligation of cell-surface receptors containing ITAM, ITIM or ITSM tyrosine phosphorylation motifs to the promiscuous cell-surface phosphatase CD452,3, which results in the direct intracellular dephosphorylation of tyrosine residues on the receptor target. As an example, we found that tonic signalling by the programmed cell death-1 receptor (PD-1) results in residual suppression of T cell activation, but is not inhibited by ligand-antagonist antibodies. We engineered a PD-1 molecule, which we denote RIPR-PD1, that induces cross-linking of PD-1 to CD45 and inhibits both tonic and ligand-activated signalling. RIPR-PD1 demonstrated enhanced inhibition of checkpoint blockade compared with ligand blocking by anti-PD1 antibodies, and increased therapeutic efficacy over anti-PD1 in mouse tumour models. We also show that the RIPR strategy extends to other immune-receptor targets that contain activating or inhibitory ITIM, ITSM or ITAM motifs; for example, inhibition of the macrophage SIRPα 'don't eat me' signal with a SIRPα-CD45 RIPR molecule potentiates antibody-dependent cellular phagocytosis beyond that of SIRPα blockade alone. RIPR represents a general strategy for direct attenuation of signalling by kinase-activated cell-surface receptors.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
