

Redox Post-translational Modifications of Protein Thiols in Brain Aging and Neurodegenerative Conditions-Focus on S-Nitrosation
- PMID: 33088270
- PMCID: PMC7497228
- DOI: 10.3389/fnagi.2020.00254
Redox Post-translational Modifications of Protein Thiols in Brain Aging and Neurodegenerative Conditions-Focus on S-Nitrosation
Abstract
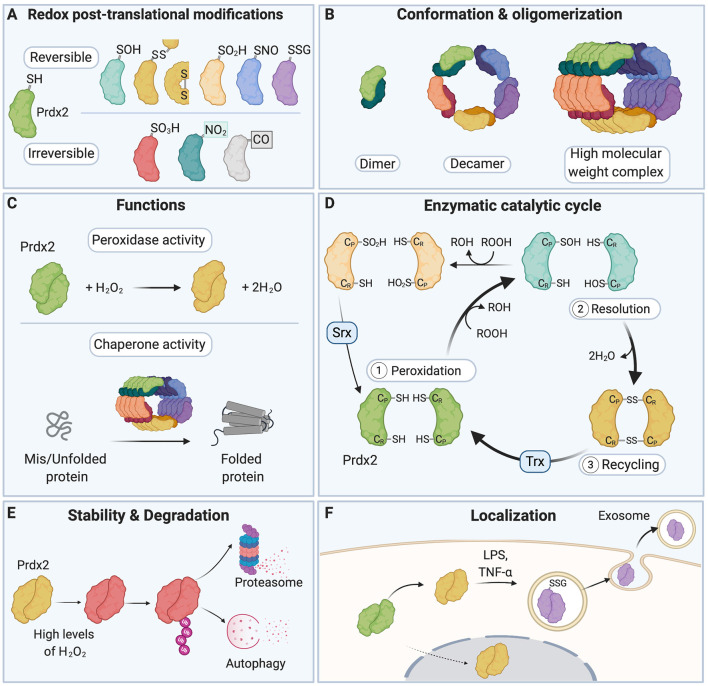
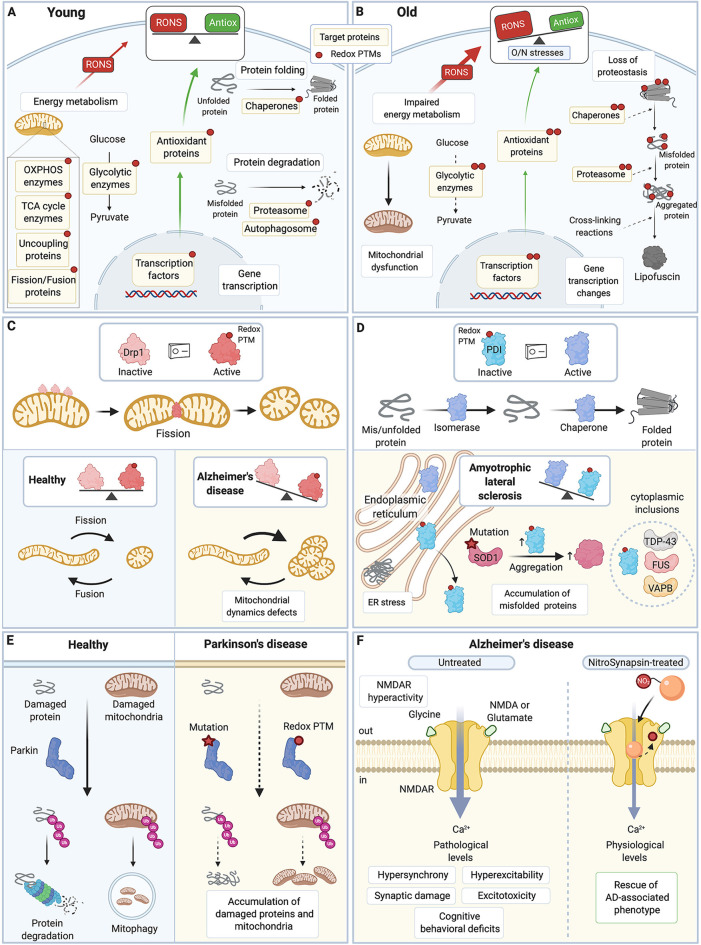
Reactive oxygen species and reactive nitrogen species (RONS) are by-products of aerobic metabolism. RONS trigger a signaling cascade that can be transduced through oxidation-reduction (redox)-based post-translational modifications (redox PTMs) of protein thiols. This redox signaling is essential for normal cellular physiology and coordinately regulates the function of redox-sensitive proteins. It plays a particularly important role in the brain, which is a major producer of RONS. Aberrant redox PTMs of protein thiols can impair protein function and are associated with several diseases. This mini review article aims to evaluate the role of redox PTMs of protein thiols, in particular S-nitrosation, in brain aging, and in neurodegenerative diseases. It also discusses the potential of using redox-based therapeutic approaches for neurodegenerative conditions.
Keywords: S-nitrosation; aging; brain; cysteine residues; neurodegenerative diseases; post-translational modifications; redox.
Copyright © 2020 Finelli.
Figures
References
-
- Ahmari M., Sharafi A., Mahmoudi J., Jafari-Anarkoli I., Gharbavi M., Hosseini M. J. (2020). Selegiline (L-Deprenyl) mitigated oxidative stress, cognitive abnormalities and histopathological change in rats: alternative therapy in transient global ischemia. J. Mol. Neurosci. [Epub ahead of print]. 10.1007/s12031-020-01544-5 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous