

Frequency-dependent selection can forecast evolution in Streptococcus pneumoniae
- PMID: 33091022
- PMCID: PMC7580979
- DOI: 10.1371/journal.pbio.3000878
Frequency-dependent selection can forecast evolution in Streptococcus pneumoniae
Abstract
Predicting how pathogen populations will change over time is challenging. Such has been the case with Streptococcus pneumoniae, an important human pathogen, and the pneumococcal conjugate vaccines (PCVs), which target only a fraction of the strains in the population. Here, we use the frequencies of accessory genes to predict changes in the pneumococcal population after vaccination, hypothesizing that these frequencies reflect negative frequency-dependent selection (NFDS) on the gene products. We find that the standardized predicted fitness of a strain, estimated by an NFDS-based model at the time the vaccine is introduced, enables us to predict whether the strain increases or decreases in prevalence following vaccination. Further, we are able to forecast the equilibrium post-vaccine population composition and assess the invasion capacity of emerging lineages. Overall, we provide a method for predicting the impact of an intervention on pneumococcal populations with potential application to other bacterial pathogens in which NFDS is a driving force.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests. ML has consulted for Pfizer, Affinivax, and Merck and has received grant support not related to this paper from Pfizer and PATH Vaccine Solutions. WPH, ML, and NJC have consulted for Antigen Discovery Inc. The authors have declared that no competing interests exist. KLOB has received grant support for pneumococcal work not related to this paper from Pfizer, GSK, and Gavi. KLOB has consulted for Merck and Sanofi Pasteur. LRG, LLH, and RCW have received grant support not related to this paper from Pfizer, Merck, and GSK.
Figures
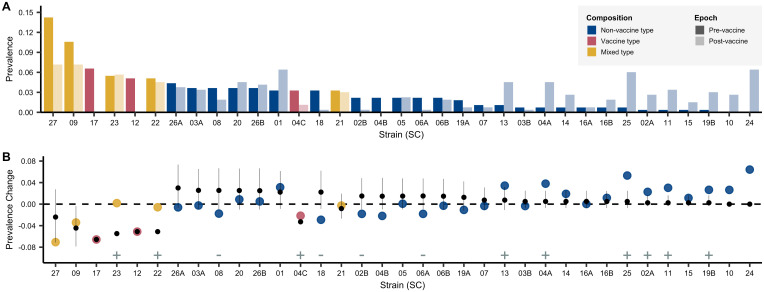
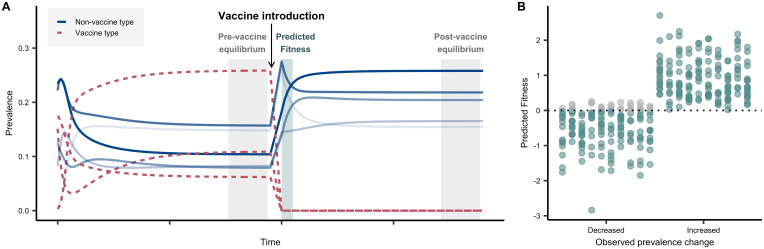
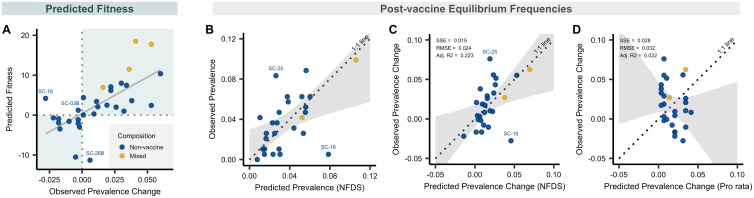
References
-
- Levin BR, Lipsitch M, Bonhoeffer S. Population Biology, Evolution, and Infectious Disease: Convergence and Synthesis. Science (80-). 1999;283:806 LP– 809. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
