

Understanding the Pathogenesis of Spondyloarthritis
- PMID: 33092023
- PMCID: PMC7588965
- DOI: 10.3390/biom10101461
Understanding the Pathogenesis of Spondyloarthritis
Abstract
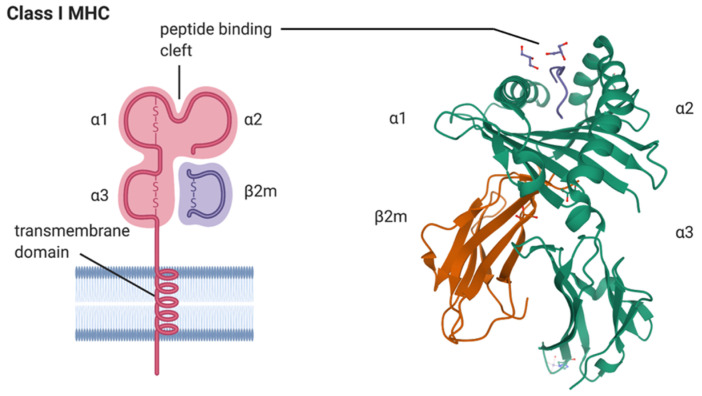
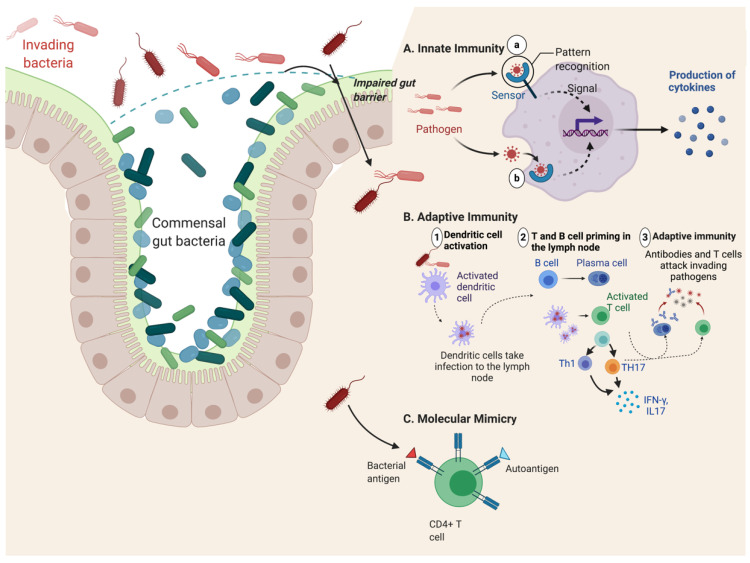
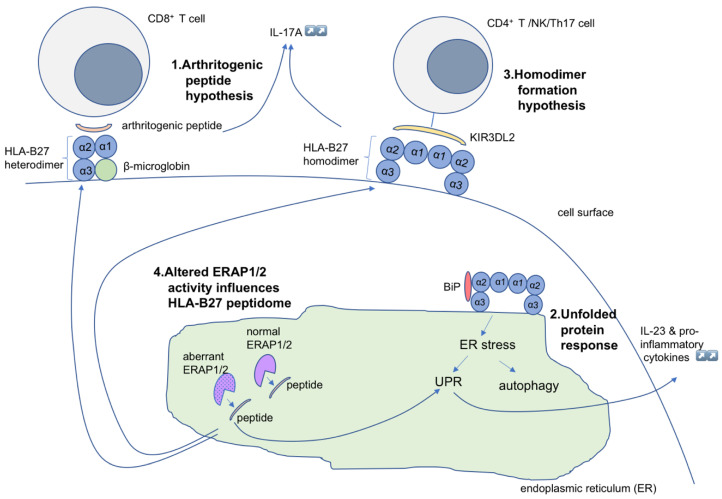
Spondyloarthritis comprises a group of inflammatory diseases of the joints and spine, with various clinical manifestations. The group includes ankylosing spondylitis, reactive arthritis, psoriatic arthritis, arthritis associated with inflammatory bowel disease, and undifferentiated spondyloarthritis. The exact etiology and pathogenesis of spondyloarthritis are still unknown, but five hypotheses explaining the pathogenesis exist. These hypotheses suggest that spondyloarthritis is caused by arthritogenic peptides, an unfolded protein response, HLA-B*27 homodimer formation, malfunctioning endoplasmic reticulum aminopeptidases, and, last but not least, gut inflammation and dysbiosis. Here we discuss the five hypotheses and the evidence supporting each. In all of these hypotheses, HLA-B*27 plays a central role. It is likely that a combination of these hypotheses, with HLA-B*27 taking center stage, will eventually explain the development of spondyloarthritis in predisposed individuals.
Keywords: ERAP1; HLA-B*27; arthritogenic peptides; gut dysbiosis; inflammation; pathogenesis; spondyloarthritis; unfolded protein response.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
