

Targeting Metabolism in Cancer Cells and the Tumour Microenvironment for Cancer Therapy
- PMID: 33092283
- PMCID: PMC7588013
- DOI: 10.3390/molecules25204831
Targeting Metabolism in Cancer Cells and the Tumour Microenvironment for Cancer Therapy
Abstract
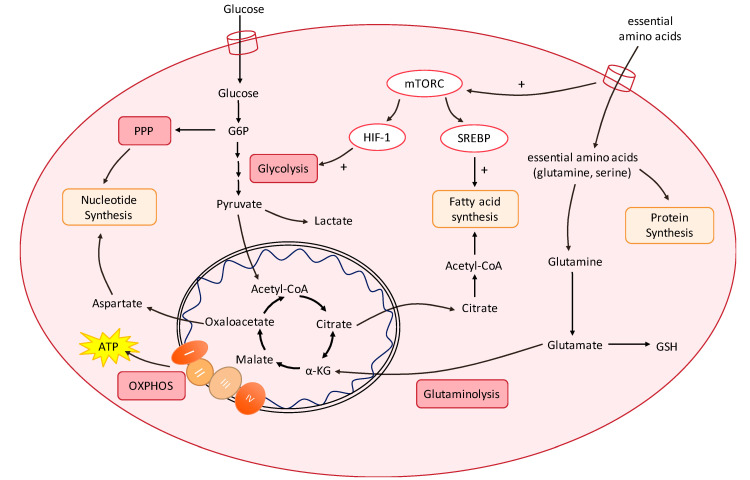
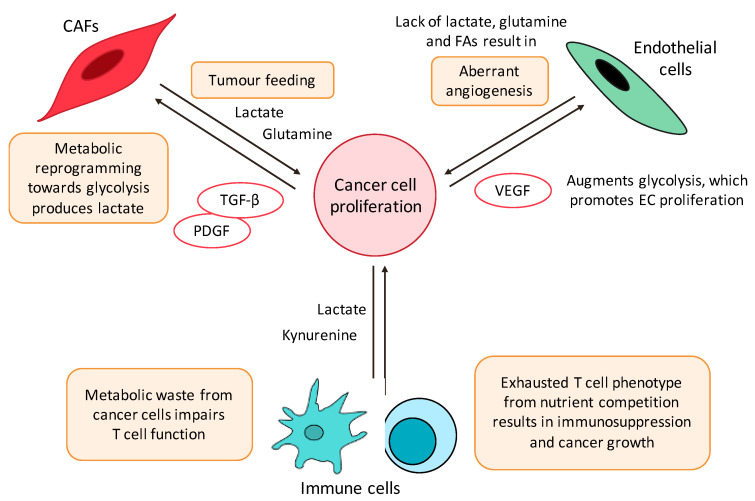
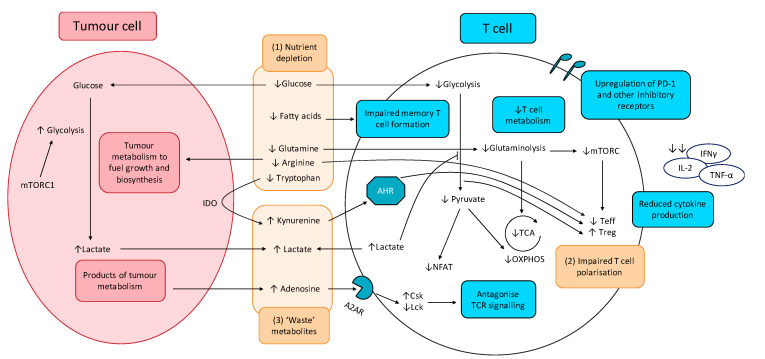
Targeting altered tumour metabolism is an emerging therapeutic strategy for cancer treatment. The metabolic reprogramming that accompanies the development of malignancy creates targetable differences between cancer cells and normal cells, which may be exploited for therapy. There is also emerging evidence regarding the role of stromal components, creating an intricate metabolic network consisting of cancer cells, cancer-associated fibroblasts, endothelial cells, immune cells, and cancer stem cells. This metabolic rewiring and crosstalk with the tumour microenvironment play a key role in cell proliferation, metastasis, and the development of treatment resistance. In this review, we will discuss therapeutic opportunities, which arise from dysregulated metabolism and metabolic crosstalk, highlighting strategies that may aid in the precision targeting of altered tumour metabolism with a focus on combinatorial therapeutic strategies.
Keywords: cancer cell metabolism; immunotherapy; metabolic reprogramming; targeted therapy; tumour microenvironment.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
