

Pyruvate dehydrogenase complex deficiency: updating the clinical, metabolic and mutational landscapes in a cohort of Portuguese patients
- PMID: 33092611
- PMCID: PMC7579914
- DOI: 10.1186/s13023-020-01586-3
Pyruvate dehydrogenase complex deficiency: updating the clinical, metabolic and mutational landscapes in a cohort of Portuguese patients
Abstract
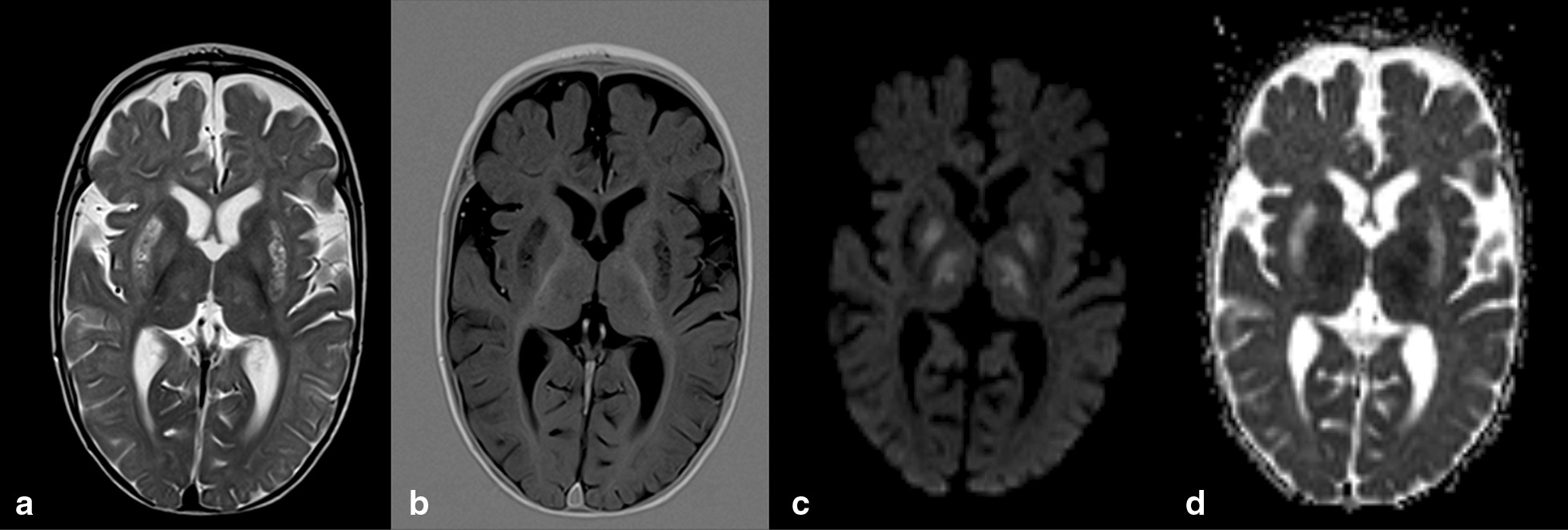
Background: The pyruvate dehydrogenase complex (PDC) catalyzes the irreversible decarboxylation of pyruvate into acetyl-CoA. PDC deficiency can be caused by alterations in any of the genes encoding its several subunits. The resulting phenotype, though very heterogeneous, mainly affects the central nervous system. The aim of this study is to describe and discuss the clinical, biochemical and genotypic information from thirteen PDC deficient patients, thus seeking to establish possible genotype-phenotype correlations.
Results: The mutational spectrum showed that seven patients carry mutations in the PDHA1 gene encoding the E1α subunit, five patients carry mutations in the PDHX gene encoding the E3 binding protein, and the remaining patient carries mutations in the DLD gene encoding the E3 subunit. These data corroborate earlier reports describing PDHA1 mutations as the predominant cause of PDC deficiency but also reveal a notable prevalence of PDHX mutations among Portuguese patients, most of them carrying what seems to be a private mutation (p.R284X). The biochemical analyses revealed high lactate and pyruvate plasma levels whereas the lactate/pyruvate ratio was below 16; enzymatic activities, when compared to control values, indicated to be independent from the genotype and ranged from 8.5% to 30%, the latter being considered a cut-off value for primary PDC deficiency. Concerning the clinical features, all patients displayed psychomotor retardation/developmental delay, the severity of which seems to correlate with the type and localization of the mutation carried by the patient. The therapeutic options essentially include the administration of a ketogenic diet and supplementation with thiamine, although arginine aspartate intake revealed to be beneficial in some patients. Moreover, in silico analysis of the missense mutations present in this PDC deficient population allowed to envisage the molecular mechanism underlying these pathogenic variants.
Conclusion: The identification of the disease-causing mutations, together with the functional and structural characterization of the mutant protein variants, allow to obtain an insight on the severity of the clinical phenotype and the selection of the most appropriate therapy.
Keywords: Genotype–phenotype correlation; Lactic acidosis; Mutational analysis; Neurological dysfunction; Pyruvate dehydrogenase complex deficiency.
Conflict of interest statement
The Authors declare they have no conflict of interest.
Figures



References
-
- Cate RL, Roche TE, Davis LC. Rapid intersite transfer of acetyl groups and movement of pyruvate dehydrogenase component in the kidney pyruvate dehydrogenase complex. J Biol Chem. 1980;255(16):7556–7562. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
