

B-cell clonogenic activity of HIV-1 p17 variants is driven by PAR1-mediated EGF transactivation
- PMID: 33093643
- PMCID: PMC8203498
- DOI: 10.1038/s41417-020-00246-9
B-cell clonogenic activity of HIV-1 p17 variants is driven by PAR1-mediated EGF transactivation
Abstract
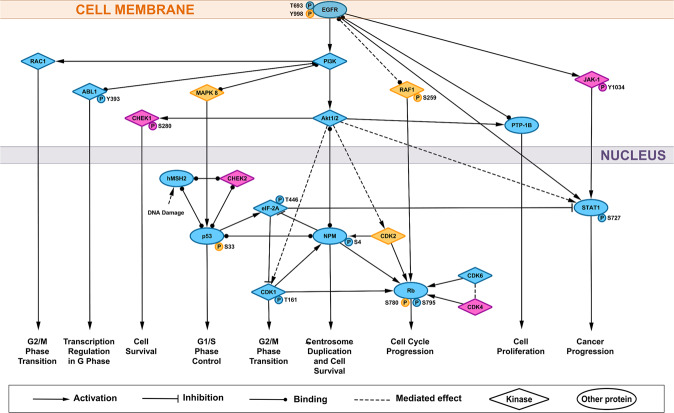
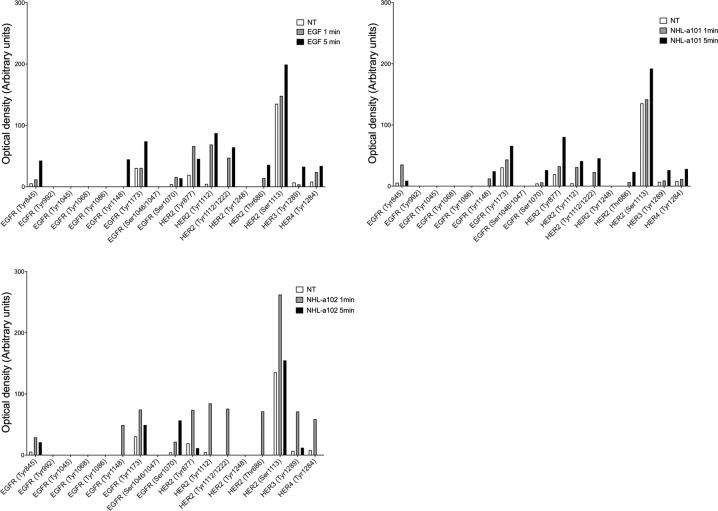
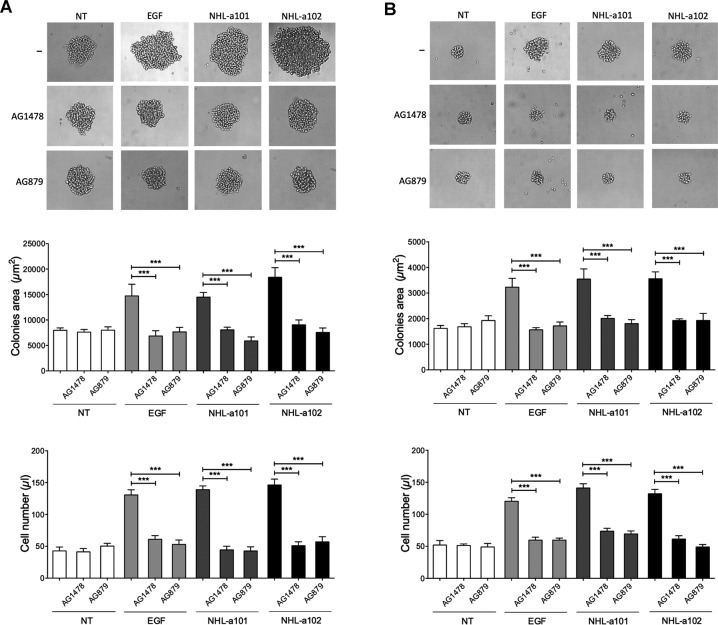
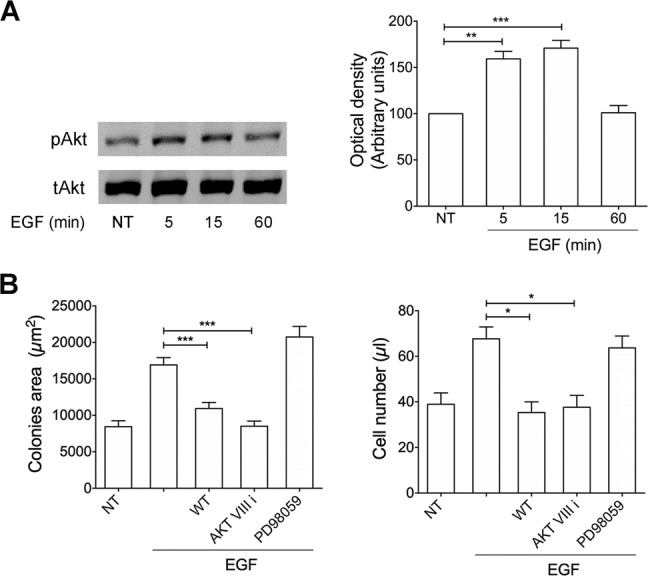
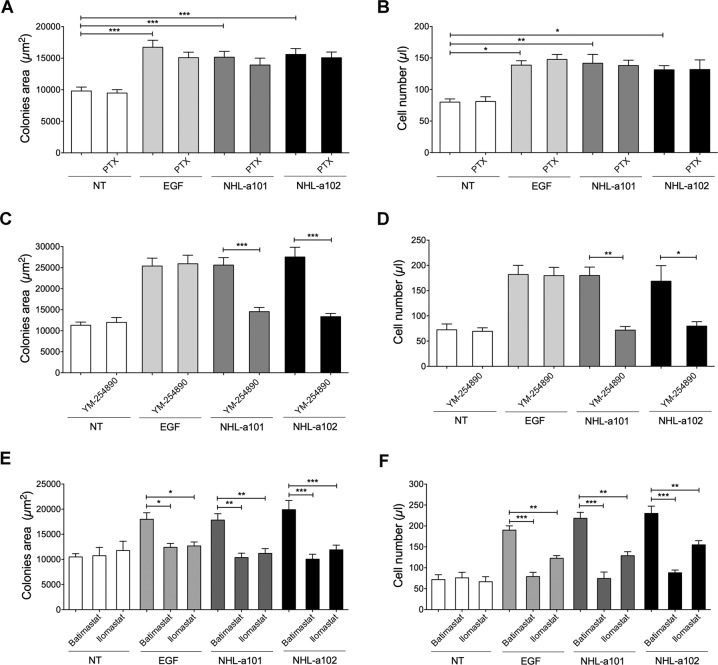
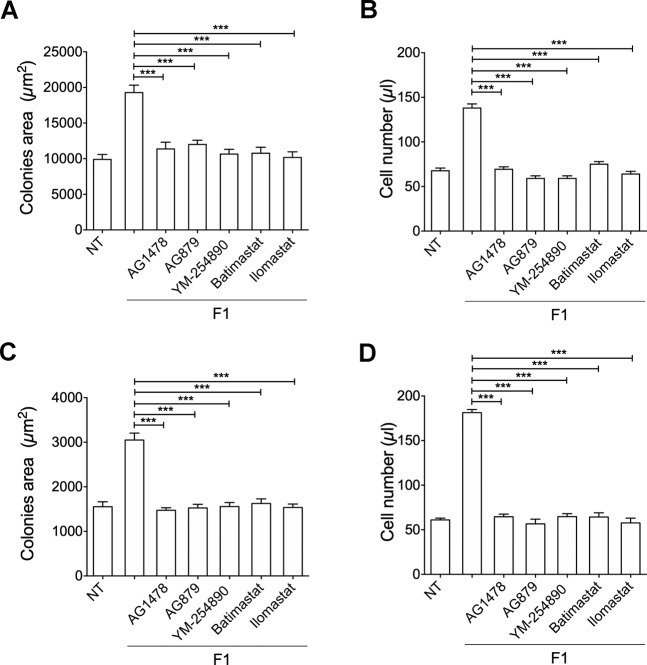
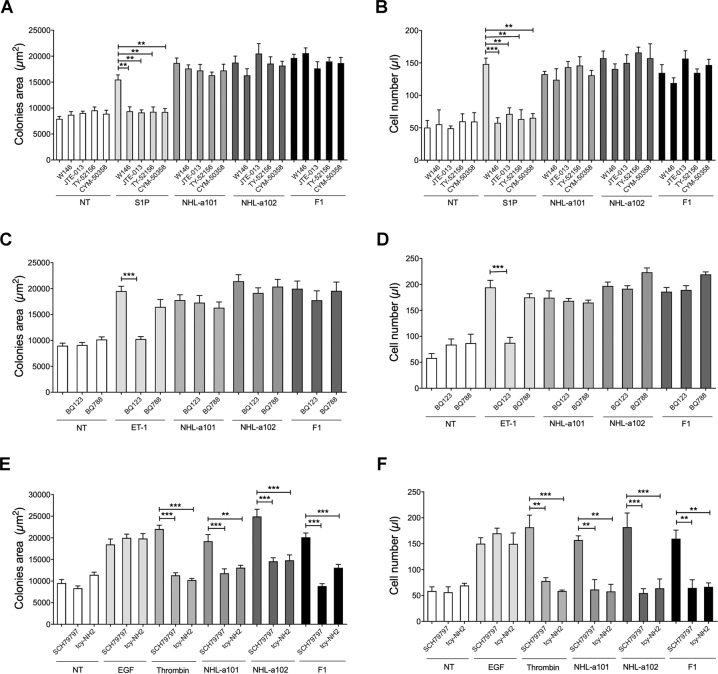
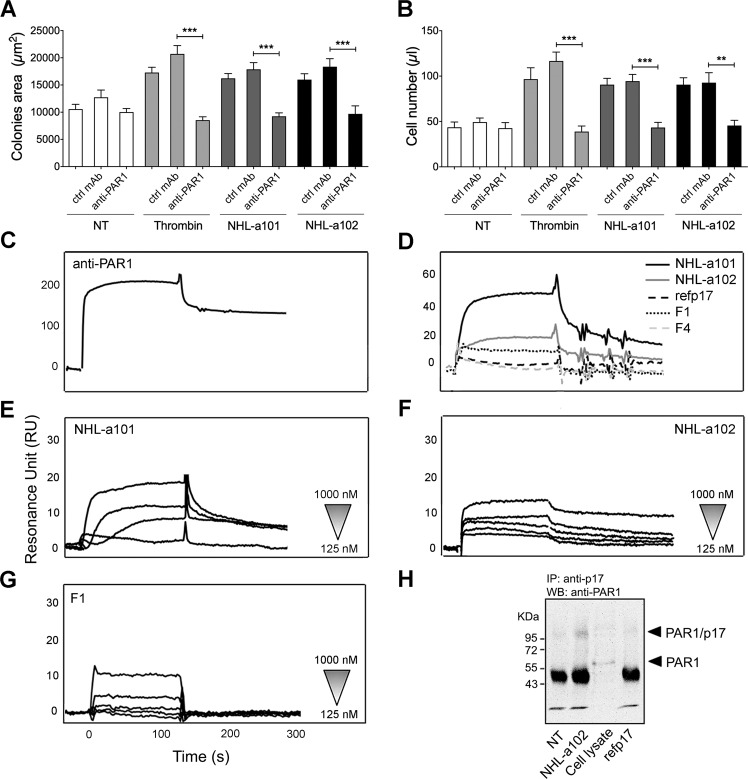
Combined antiretroviral therapy (cART) for HIV-1 dramatically slows disease progression among HIV+ individuals. Currently, lymphoma represents the main cause of death among HIV-1-infected patients. Detection of p17 variants (vp17s) endowed with B-cell clonogenic activity in HIV-1-seropositive patients with lymphoma suggests their possible role in lymphomagenesis. Here, we demonstrate that the clonogenic activity of vp17s is mediated by their binding to PAR1 and to PAR1-mediated EGFR transactivation through Gq protein. The entire vp17s-triggered clonogenic process is MMPs dependent. Moreover, phosphoproteomic and bioinformatic analysis highlighted the crucial role of EGFR/PI3K/Akt pathway in modulating several molecules promoting cancer progression, including RAC1, ABL1, p53, CDK1, NPM, Rb, PTP-1B, and STAT1. Finally, we show that a peptide (F1) corresponding to the vp17s functional epitope is sufficient to trigger the PAR1/EGFR/PI3K/Akt pathway and bind PAR1. Our findings suggest novel potential therapeutic targets to counteract vp17-driven lymphomagenesis in HIV+ patients.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Rabkin C. Epidemiology of AIDS‐related malignancies. Curr Opin Oncol. 1994;6:492–6. - PubMed
-
- He B, Qiao X, Klasse PJ, Chiu A, Chadburn A, Knowles DM, et al. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J Immunol. 2006;176:3931–41. - PubMed
-
- Kundu RK, Sangiorgi F, Wu LY, Pattengale PK, Hinton DR, Gill PS, et al. Expression of the human immunodeficiency virus-Tat gene in lymphoid tissues of transgenic mice is associated with B-cell lymphoma. Blood. 1999;94:275–82. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
