

Novel insights into the epidemiology of epidermolysis bullosa (EB) from the Dutch EB Registry: EB more common than previously assumed?
- PMID: 33095945
- PMCID: PMC7984089
- DOI: 10.1111/jdv.17012
Novel insights into the epidemiology of epidermolysis bullosa (EB) from the Dutch EB Registry: EB more common than previously assumed?
Abstract
Background: Epidermolysis bullosa (EB) is a heterogeneous group of rare and incurable genetic disorders characterized by fragility of the skin and mucosae, resulting in blisters and erosions. Several epidemiological studies in other populations have been carried out, reporting varying and sometimes inconclusive figures, highlighting the need for standardized epidemiological analyses in well-characterized cohorts.
Objectives: To evaluate the epidemiological data on EB in the Netherlands, extracted from the molecularly well-characterized cohort in the Dutch EB Registry.
Methods: In this observational study all EB-patients that were based in the Netherlands and captured in the Dutch EB Registry between 1988 and 2018 were included. The epidemiological outcomes were based on complete diagnostic data (clinical features, immunofluorescence, electron microscopy and mutation analysis), with longitudinal follow-up.
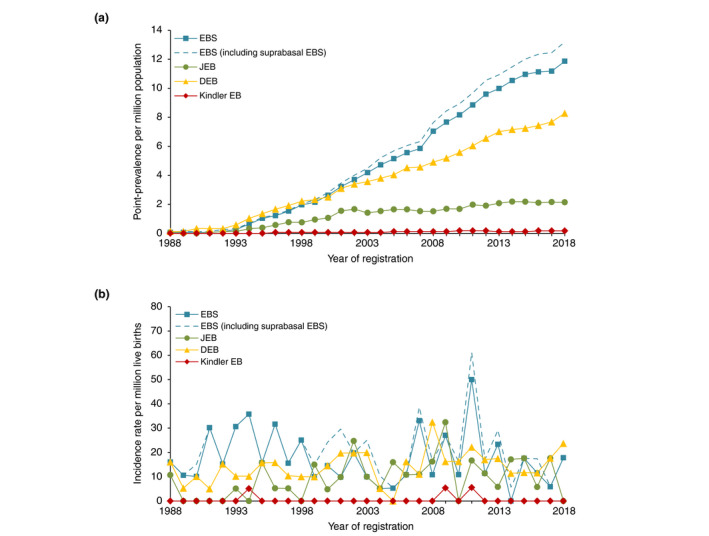
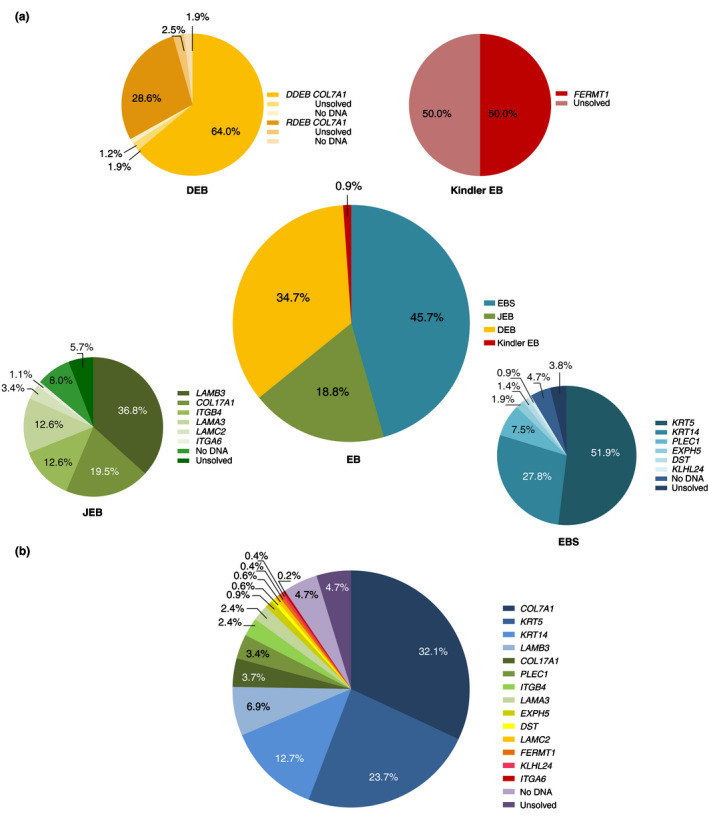
Results: A total of 464 EB-patients (287 families) were included. The incidence and point-prevalence of EB in the Netherlands were 41.3 per million live births and 22.4 per million population, respectively. EB Simplex (EBS), Junctional EB (JEB), Dystrophic EB (DEB) and Kindler EB were diagnosed in 45.7%, 18.8%, 34.7% and 0.9% of the EB-patients, respectively, with an incidence and point-prevalence of 17.5 and 11.9 (EBS), 9.3 and 2.1 (JEB), 14.1 and 8.3 (DEB), 0.5 and 0.2 (Kindler EB). In 90.5% of the EB-patients the diagnosis was genetically confirmed. During the investigated time period 73 EB-patients died, 72.6% of whom as a direct consequence of their EB.
Conclusion: The epidemiological outcomes of EB in the Netherlands are high, attributed to a high detection rate in a well-organized set-up, indicating that EB might be more common than previously assumed. These epidemiological data help to understand the extensive need for (specialized) medical care of EB-patients and is invaluable for the design and execution of therapeutic trials. This study emphasizes the importance of thorough reporting systems and registries worldwide.
© 2020 The Authors. Journal of the European Academy of Dermatology and Venereology published by John Wiley & Sons Ltd on behalf of European Academy of Dermatology and Venereology.
Figures
References
-
- Fine JD, Bruckner‐Tuderman L, Eady RA et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70: 1103–1126. - PubMed
-
- Has C, Bauer JW, Bodemer C et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol 2020; 183: 614–627. - PubMed
-
- Duipmans JC, Jonkman MF. Interdisciplinary management of epidermolysis bullosa in the public setting: the Netherlands as a model of care. Dermatol Clin 2010; 28: 383–386. - PubMed
-
- Turcan I, Pasmooij AMG, van den Akker PC et al. Heterozygosity for a novel missense mutation in the ITGB4 gene associated with autosomal dominant epidermolysis bullosa. JAMA Dermatol 2016; 152: 558–562. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
