

Predicting Drug Response and Synergy Using a Deep Learning Model of Human Cancer Cells
- PMID: 33096023
- PMCID: PMC7737474
- DOI: 10.1016/j.ccell.2020.09.014
Predicting Drug Response and Synergy Using a Deep Learning Model of Human Cancer Cells
Abstract
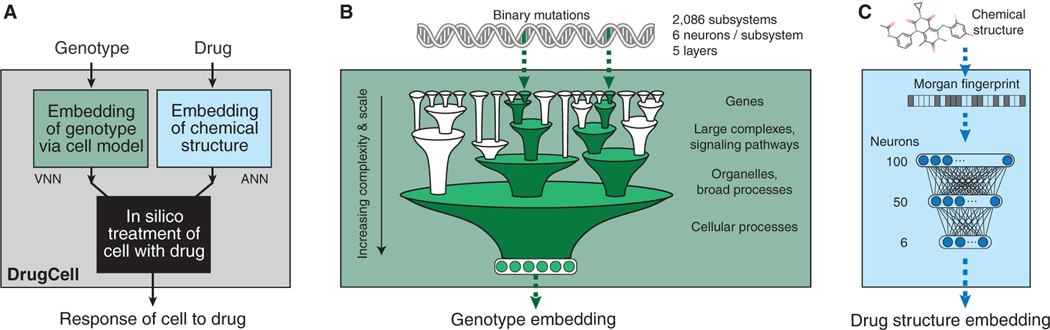
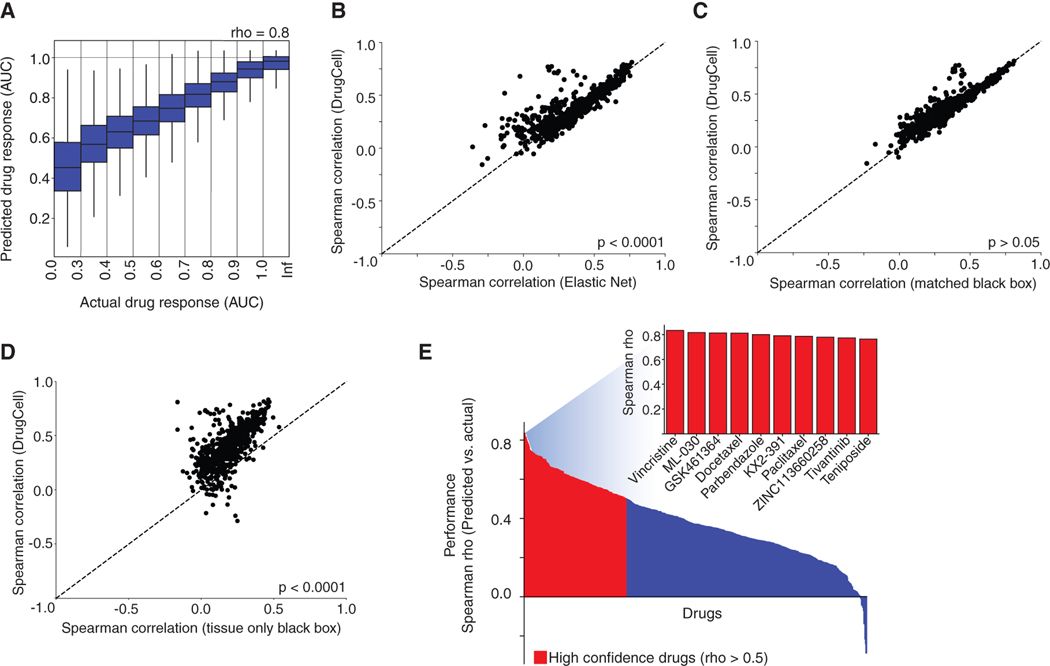
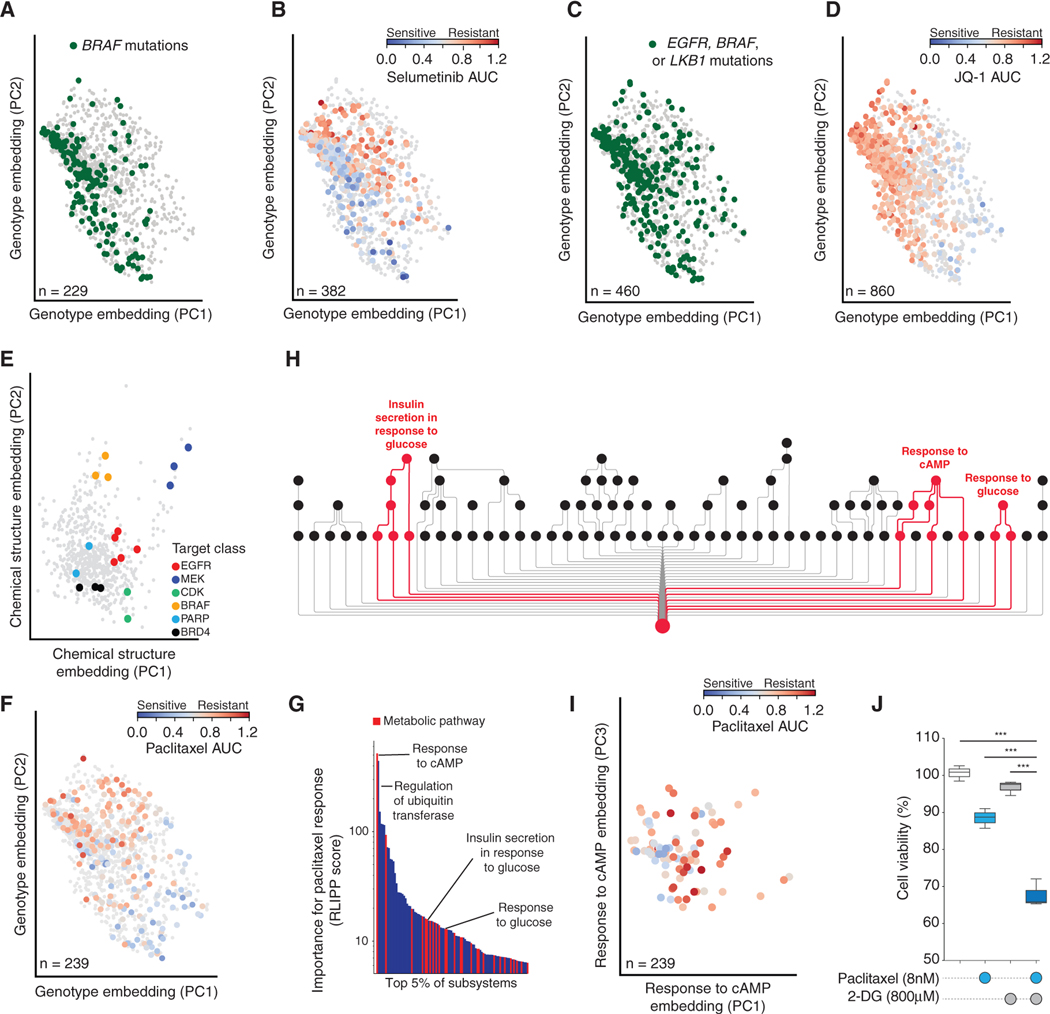
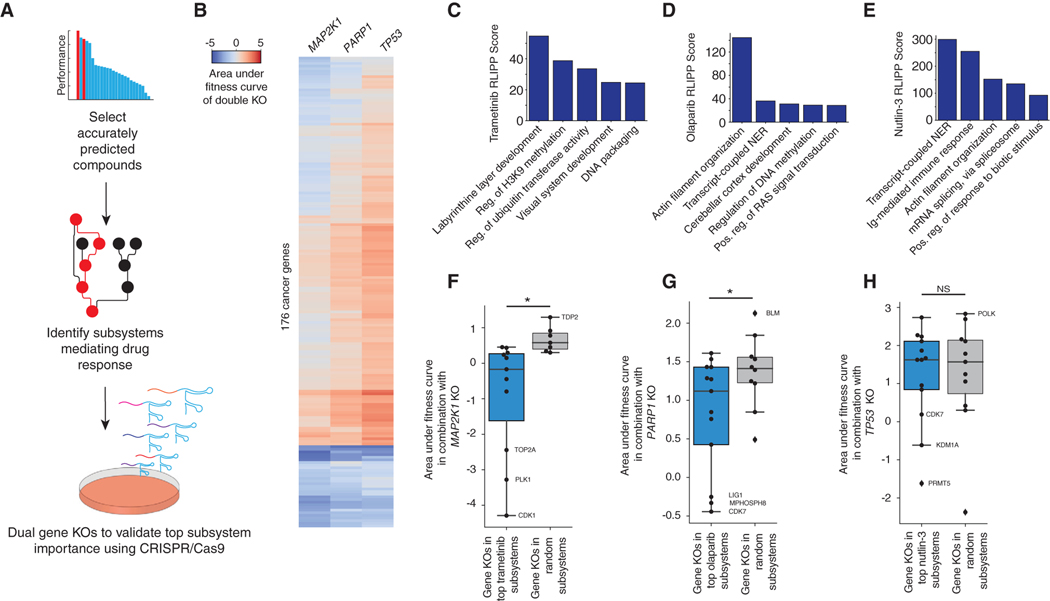
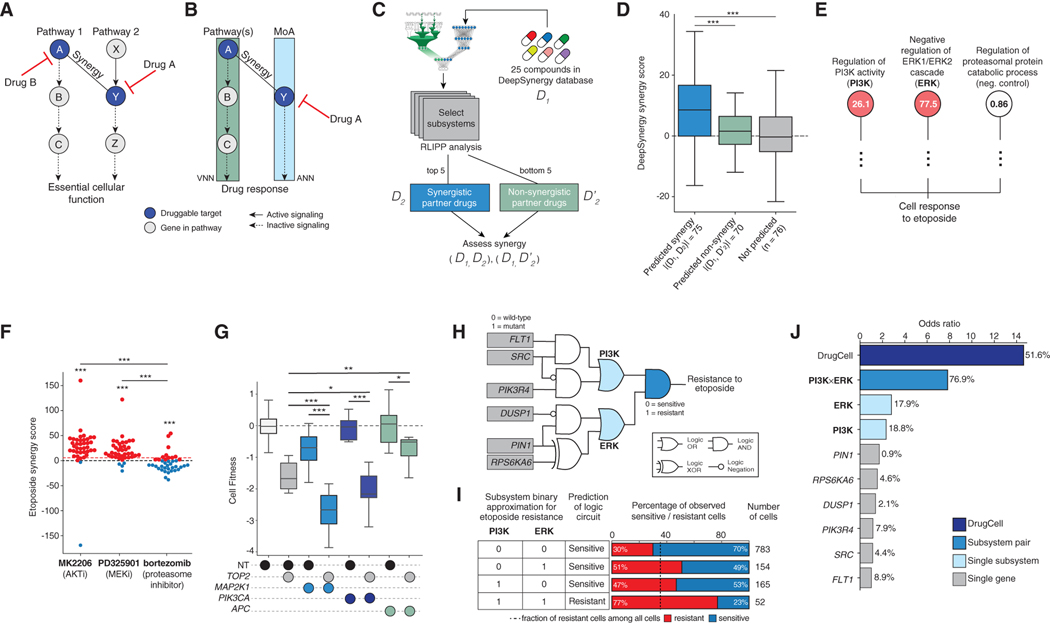
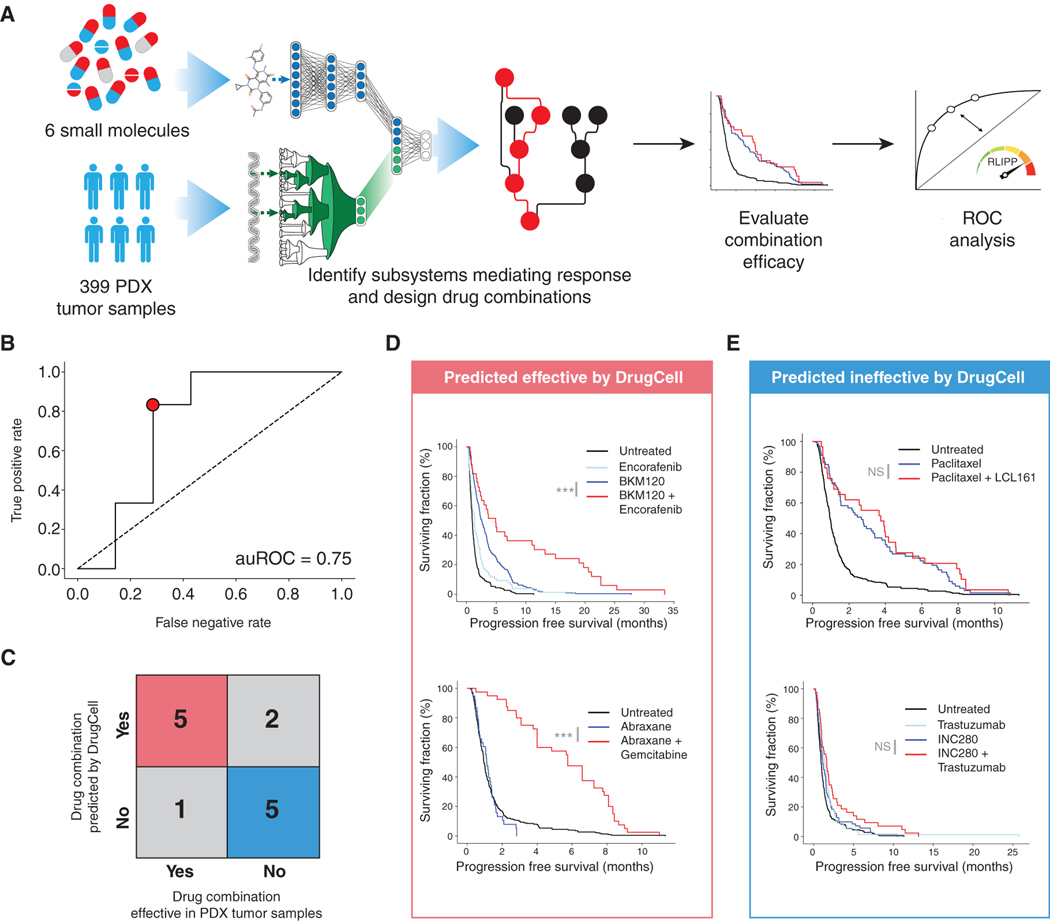
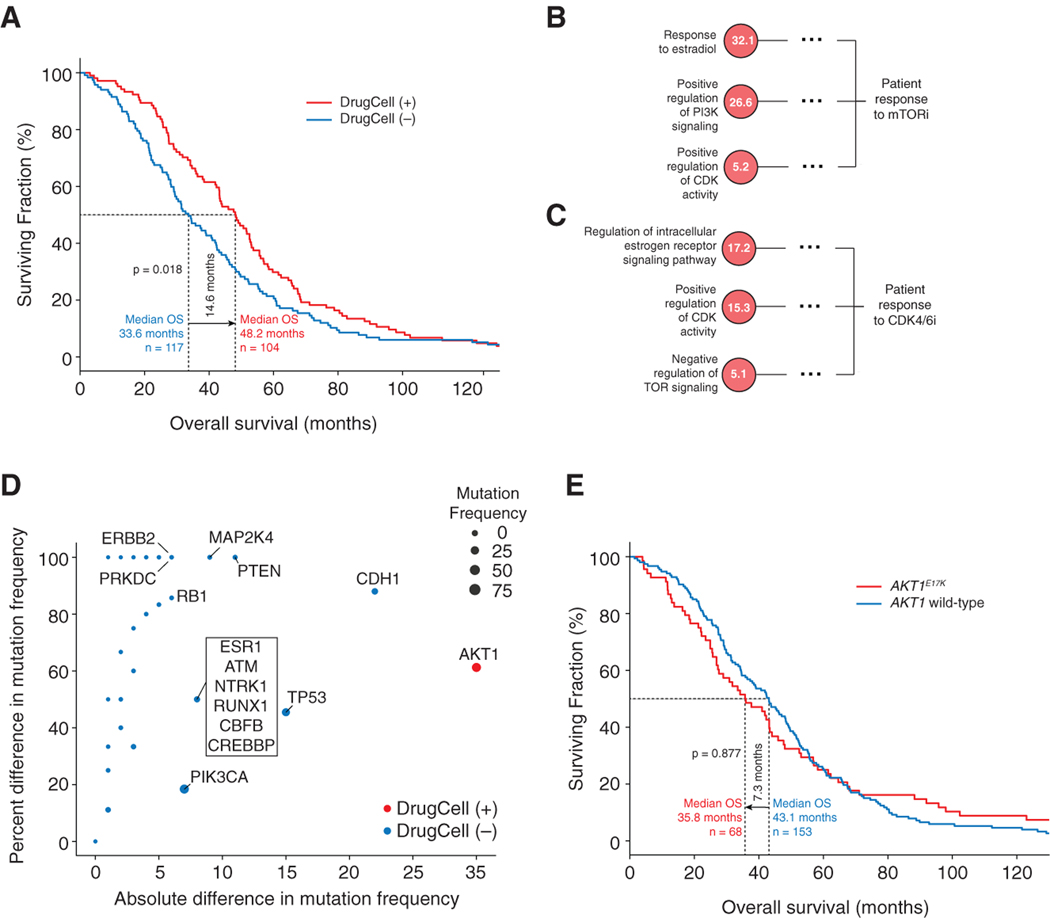
Most drugs entering clinical trials fail, often related to an incomplete understanding of the mechanisms governing drug response. Machine learning techniques hold immense promise for better drug response predictions, but most have not reached clinical practice due to their lack of interpretability and their focus on monotherapies. We address these challenges by developing DrugCell, an interpretable deep learning model of human cancer cells trained on the responses of 1,235 tumor cell lines to 684 drugs. Tumor genotypes induce states in cellular subsystems that are integrated with drug structure to predict response to therapy and, simultaneously, learn biological mechanisms underlying the drug response. DrugCell predictions are accurate in cell lines and also stratify clinical outcomes. Analysis of DrugCell mechanisms leads directly to the design of synergistic drug combinations, which we validate systematically by combinatorial CRISPR, drug-drug screening in vitro, and patient-derived xenografts. DrugCell provides a blueprint for constructing interpretable models for predictive medicine.
Keywords: cancer; drug synergy; interpretable deep learning; machine learning; network modeling; precision medicine.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests T.I. is a co-founder of Data4Cure, Inc., and has an equity interest. T.I. has an equity interest in Ideaya BioSciences, Inc. The terms of this arrangement have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies.
Figures
Comment in
-
Biologically Informed Neural Networks Predict Drug Responses.Cancer Cell. 2020 Nov 9;38(5):613-615. doi: 10.1016/j.ccell.2020.10.014. Epub 2020 Oct 22. Cancer Cell. 2020. PMID: 33096022
-
Artificial Intelligence Predicts Drug Response.Cancer Discov. 2021 Jan;11(1):4-5. doi: 10.1158/2159-8290.CD-NB2020-109. Epub 2020 Nov 25. Cancer Discov. 2021. PMID: 33239267
References
-
- Bani MR, Nicoletti MI, Alkharouf NW, Ghilardi C, Petersen D, Erba E, Sausville EA, Liu ET, and Giavazzi R (2004). Gene expression correlating with response to paclitaxel in ovarian carcinoma xenografts. Mol. Cancer Ther 3, 111–121. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
