Phenotypic and Transcriptomic Analyses of Seven Clinical Stenotrophomonas maltophilia Isolates Identify a Small Set of Shared and Commonly Regulated Genes Involved in the Biofilm Lifestyle
- PMID: 33097507
- PMCID: PMC7688217
- DOI: 10.1128/AEM.02038-20
Phenotypic and Transcriptomic Analyses of Seven Clinical Stenotrophomonas maltophilia Isolates Identify a Small Set of Shared and Commonly Regulated Genes Involved in the Biofilm Lifestyle
Erratum in
-
Erratum for Alio et al., "Phenotypic and Transcriptomic Analyses of Seven Clinical Stenotrophomonas maltophilia Isolates Identify a Small Set of Shared and Commonly Regulated Genes Involved in the Biofilm Lifestyle".Appl Environ Microbiol. 2021 Aug 26;87(18):e0130421. doi: 10.1128/AEM.01304-21. Epub 2021 Aug 26. Appl Environ Microbiol. 2021. PMID: 34435871 Free PMC article. No abstract available.
Abstract
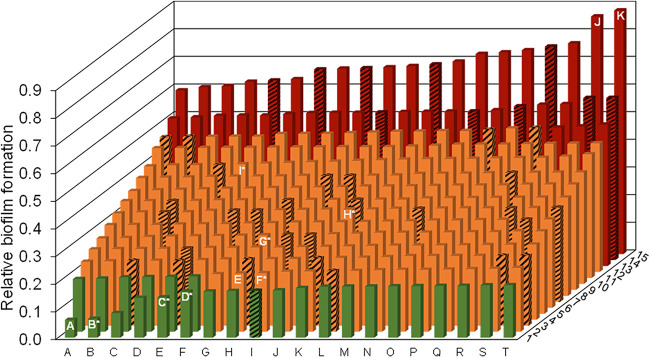
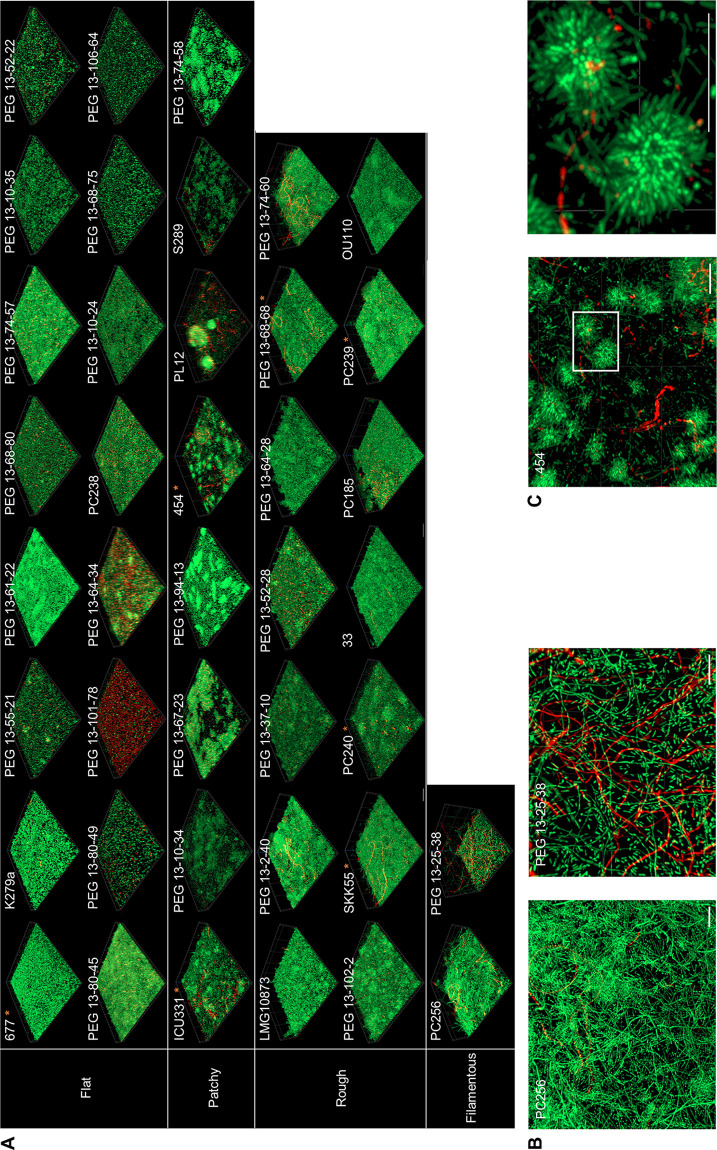
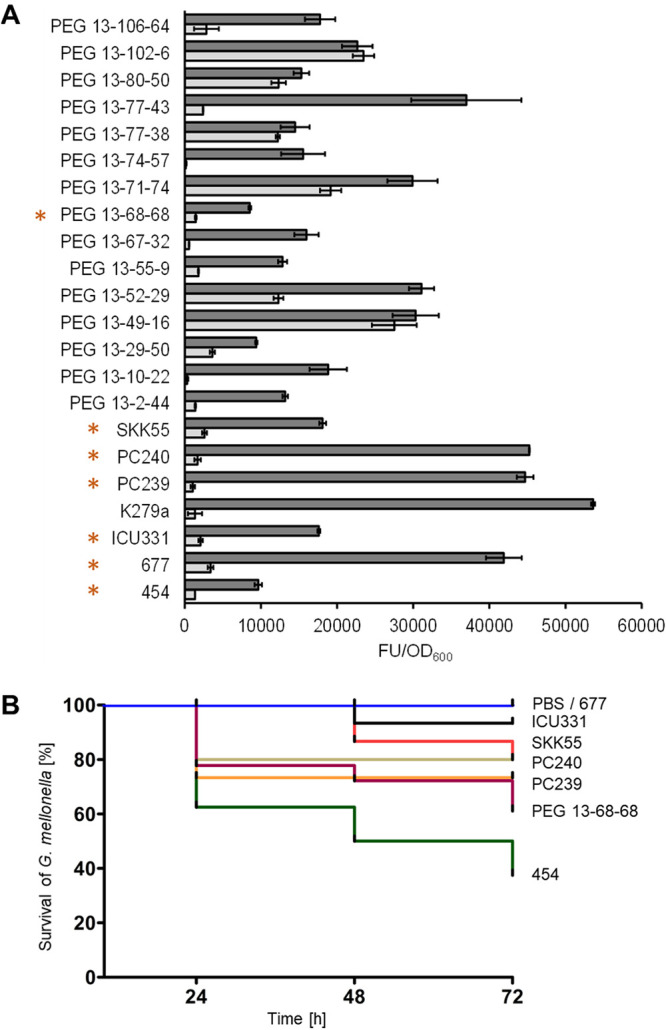
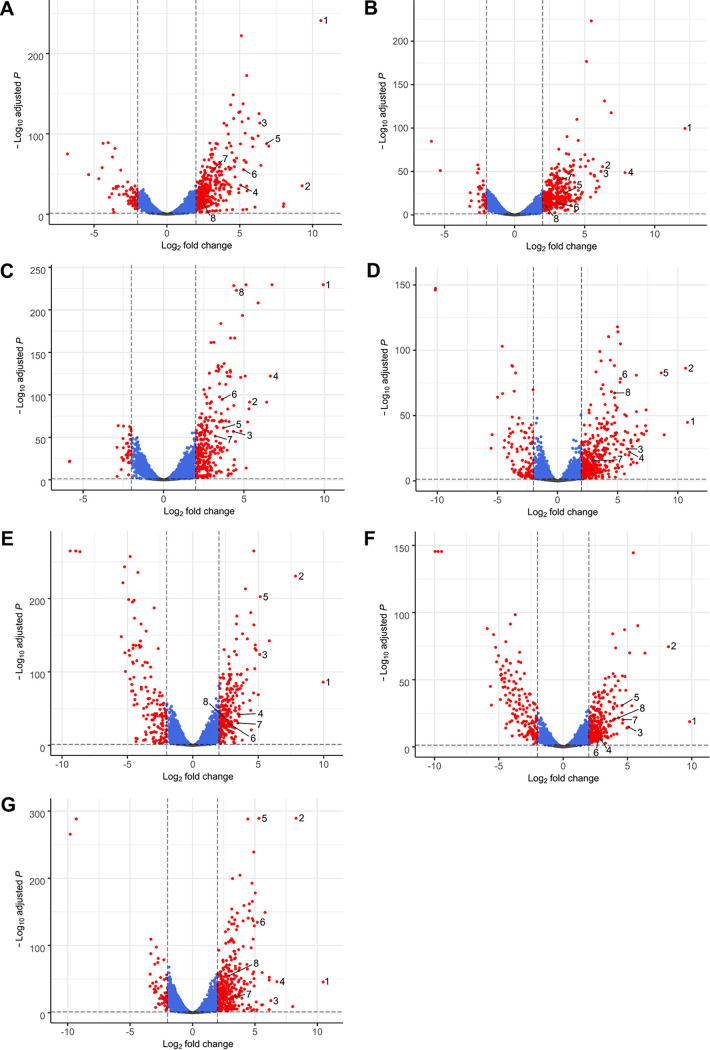
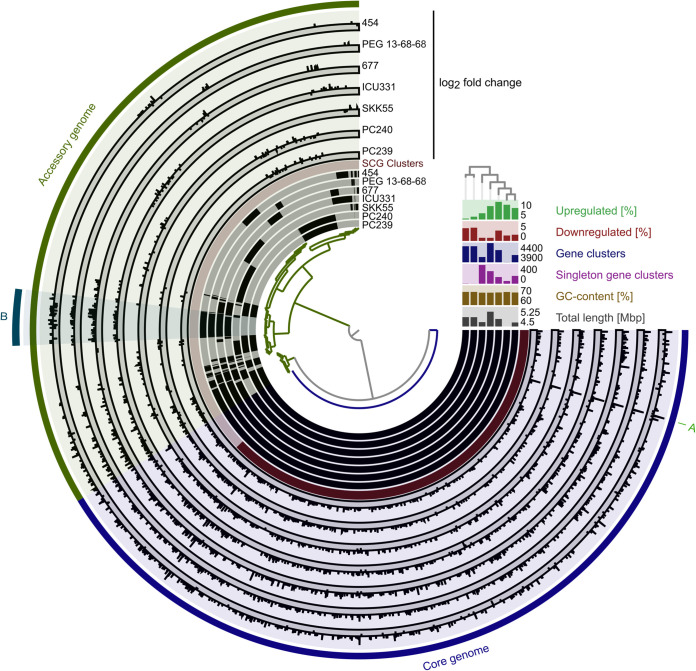
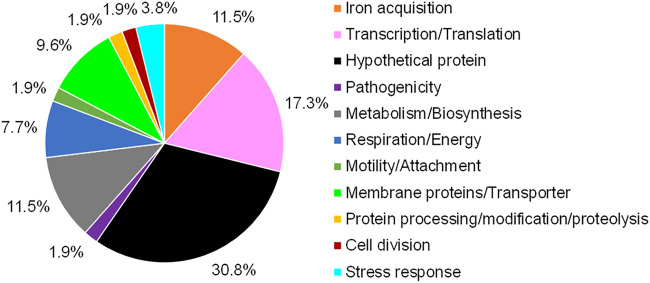
Stenotrophomonas maltophilia is one of the most frequently isolated multidrug-resistant nosocomial opportunistic pathogens. It contributes to disease progression in cystic fibrosis (CF) patients and is frequently isolated from wounds, infected tissues, and catheter surfaces. On these diverse surfaces S. maltophilia lives in single-species or multispecies biofilms. Since very little is known about common processes in biofilms of different S. maltophilia isolates, we analyzed the biofilm profiles of 300 clinical and environmental isolates from Europe of the recently identified main lineages Sgn3, Sgn4, and Sm2 to Sm18. The analysis of the biofilm architecture of 40 clinical isolates revealed the presence of multicellular structures and high phenotypic variability at a strain-specific level. Further, transcriptome analyses of biofilm cells of seven clinical isolates identified a set of 106 shared strongly expressed genes and 33 strain-specifically expressed genes. Surprisingly, the transcriptome profiles of biofilm versus planktonic cells revealed that just 9.43% ± 1.36% of all genes were differentially regulated. This implies that just a small set of shared and commonly regulated genes is involved in the biofilm lifestyle. Strikingly, iron uptake appears to be a key factor involved in this metabolic shift. Further, metabolic analyses implied that S. maltophilia employs a mostly fermentative growth mode under biofilm conditions. The transcriptome data of this study together with the phenotypic and metabolic analyses represent so far the largest data set on S. maltophilia biofilm versus planktonic cells. This study will lay the foundation for the identification of strategies for fighting S. maltophilia biofilms in clinical and industrial settings.IMPORTANCE Microorganisms living in a biofilm are much more tolerant to antibiotics and antimicrobial substances than planktonic cells are. Thus, the treatment of infections caused by microorganisms living in biofilms is extremely difficult. Nosocomial infections (among others) caused by S. maltophilia, particularly lung infection among CF patients, have increased in prevalence in recent years. The intrinsic multidrug resistance of S. maltophilia and the increased tolerance to antimicrobial agents of its biofilm cells make the treatment of S. maltophilia infection difficult. The significance of our research is based on understanding the common mechanisms involved in biofilm formation of different S. maltophilia isolates, understanding the diversity of biofilm architectures among strains of this species, and identifying the differently regulated processes in biofilm versus planktonic cells. These results will lay the foundation for the treatment of S. maltophilia biofilms.
Keywords: Stenotrophomonas; biofilms; transcriptome.
Copyright © 2020 Alio et al.
Figures