

ATM-Dependent Recruitment of BRD7 is required for Transcriptional Repression and DNA Repair at DNA Breaks Flanking Transcriptional Active Regions
- PMID: 33101843
- PMCID: PMC7578904
- DOI: 10.1002/advs.202000157
ATM-Dependent Recruitment of BRD7 is required for Transcriptional Repression and DNA Repair at DNA Breaks Flanking Transcriptional Active Regions
Abstract
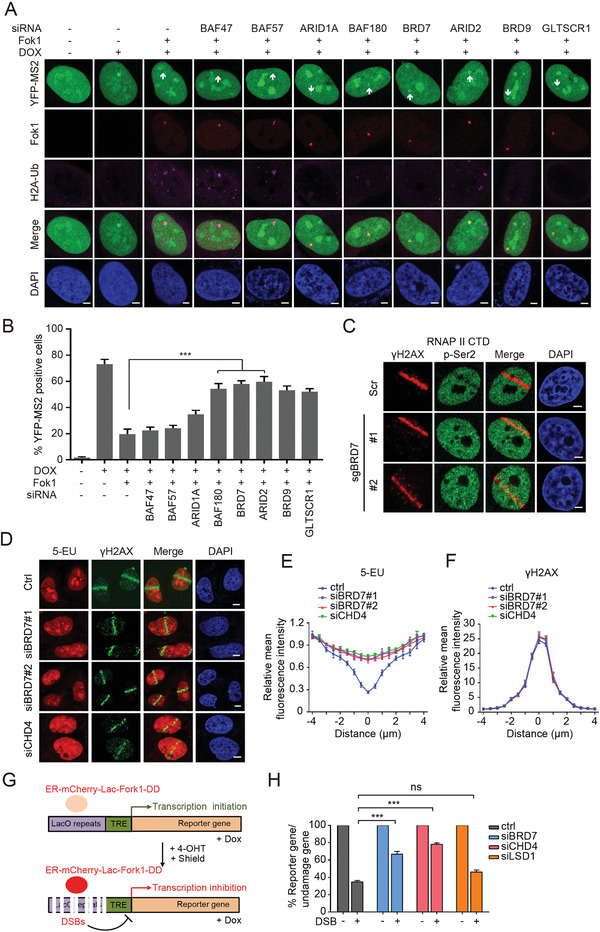
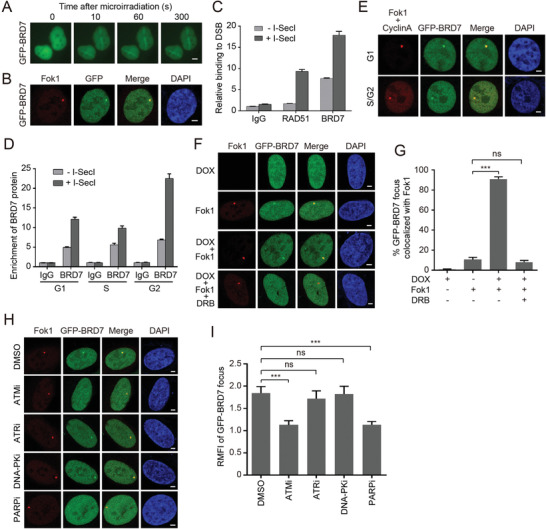
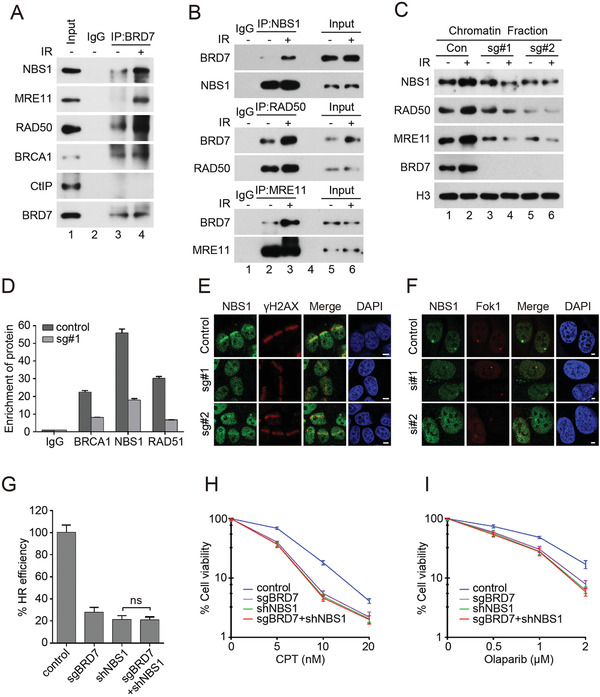
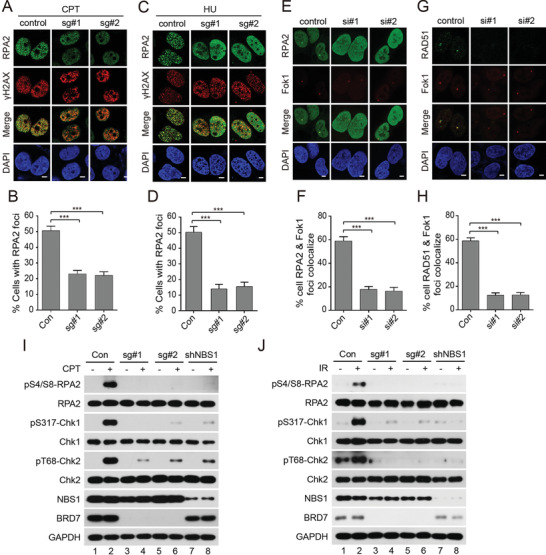
Repair of DNA double-strand breaks (DSBs) is essential for genome integrity, and is accompanied by transcriptional repression at the DSB regions. However, the mechanisms how DNA repair induces transcriptional inhibition remain elusive. Here, it is identified that BRD7 participates in DNA damage response (DDR) and is recruited to the damaged chromatin via ATM signaling. Mechanistically, BRD7 joins the polycomb repressive complex 2 (PRC2), the nucleosome remodeling and histone deacetylation (NuRD) complex at the damaged DNA and recruits E3 ubiquitin ligase RNF168 to the DSBs. Furthermore, ATM-mediated BRD7 phosphorylation is required for recruitment of the PRC2 complex, NuRD complex, DSB sensor complex MRE11-RAD50-NBS1 (MRN), and RNF168 to the active transcription sites at DSBs, resulting in transcriptional repression and DNA repair. Moreover, BRD7 deficiency sensitizes cancer cells to PARP inhibition. Collectively, BRD7 is crucial for DNA repair and DDR-mediated transcription repression, which may serve as a therapeutic target. The findings identify the missing link between DNA repair and transcription regulation that maintains genome integrity.
Keywords: ATM; BRD7; NuRD; PRC2; transcriptional repression.
© 2020 The Authors. Published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous