

Complement in Secondary Thrombotic Microangiopathy
- PMID: 33102952
- PMCID: PMC7575444
- DOI: 10.1016/j.ekir.2020.10.009
Complement in Secondary Thrombotic Microangiopathy
Abstract
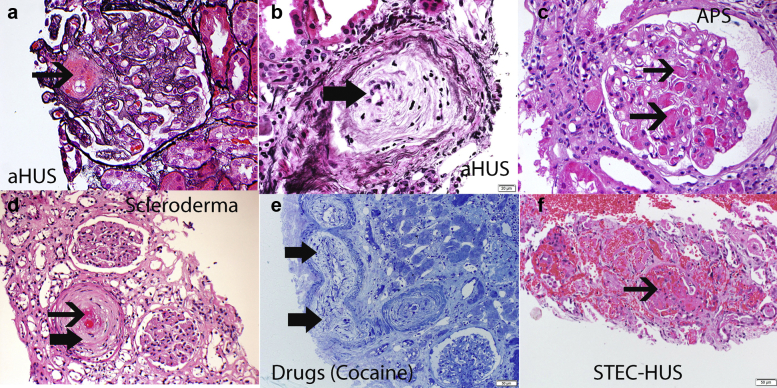
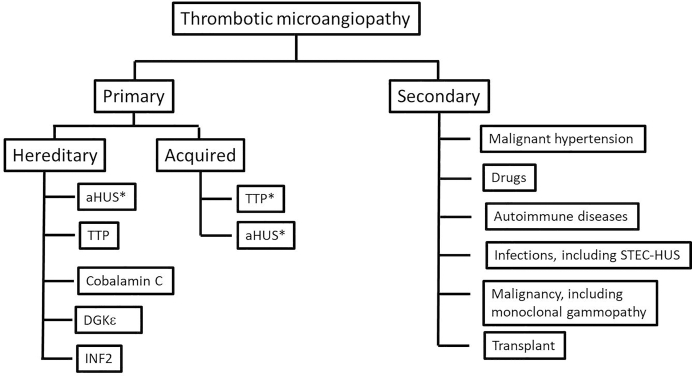
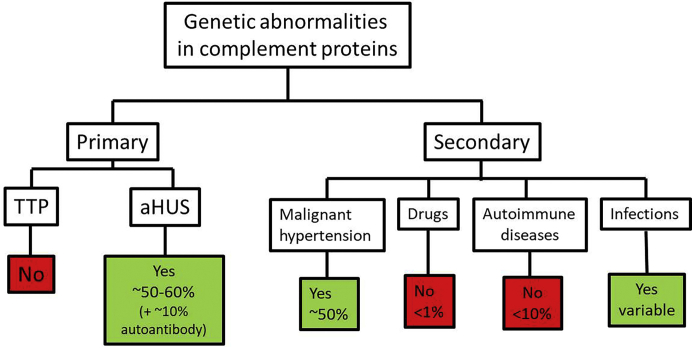
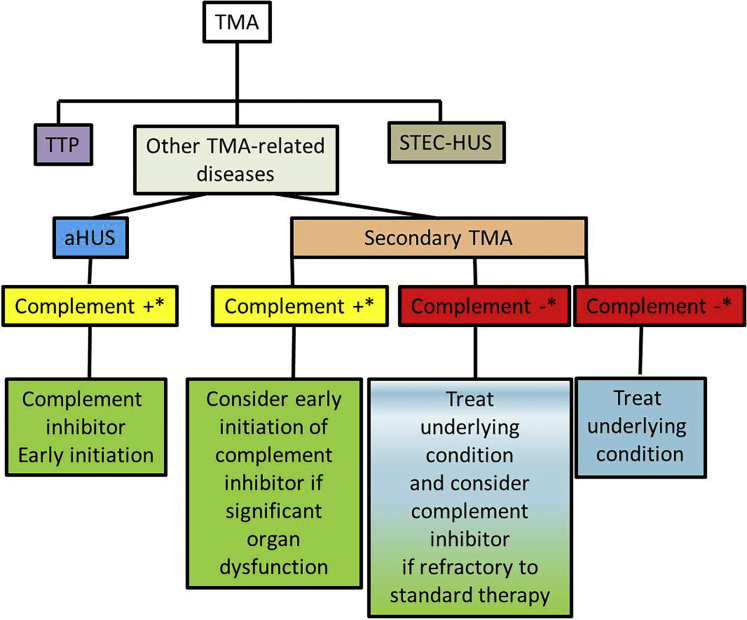
Thrombotic microangiopathy (TMA) is a condition characterized by thrombocytopenia and microangiopathic hemolytic anemia (MAHA) with varying degrees of organ damage in the setting of normal international normalized ratio and activated partial thromboplastin time. Complement has been implicated in the etiology of TMA, which are classified as primary TMA when genetic and acquired defects in complement proteins are the primary drivers of TMA (complement-mediated TMA or atypical hemolytic uremic syndrome, aHUS) or secondary TMA, when complement activation occurs in the context of other disease processes, such as infection, malignant hypertension, autoimmune disease, malignancy, transplantation, pregnancy, and drugs. It is important to recognize that this classification is not absolute because genetic variants in complement genes have been identified in patients with secondary TMA, and distinguishing complement/genetic-mediated TMA from secondary causes of TMA can be challenging and lead to potentially harmful delays in treatment. In this review, we focus on data supporting the involvement of complement in aHUS and in secondary forms of TMA associated with malignant hypertension, drugs, autoimmune diseases, pregnancy, and infections. In aHUS, genetic variants in complement genes are found in up to 60% of patients, whereas in the secondary forms, the finding of genetic defects is variable, ranging from almost 60% in TMA associated with malignant hypertension to less than 10% in drug-induced TMA. On the basis of these findings, a new approach to management of TMA is proposed.
Keywords: HUS; autoimmune disease; complement; drugs; hypertension; thrombotic microangiopathy.
© 2020 International Society of Nephrology. Published by Elsevier Inc.
Figures
References
-
- Moake J.L. Thrombotic microangiopathies. N Engl J Med. 2002;347:589–600. - PubMed
-
- Laurence J., Haller H., Mannucci P.M. Atypical hemolytic uremic syndrome (aHUS): essential aspects of an accurate diagnosis. Clin Adv Hematol Oncol. 2016;14(suppl 11):2–15. - PubMed
-
- George J.N., Nester C.M. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371:1847–1848. - PubMed
-
- Scully M., Cataland S., Coppo P. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15:312–322. - PubMed
-
- Fox L.C., Cohney S.J., Kausman J.Y., Shortt J., Hughes P.D., Wood E.M. Consensus opinion on diagnosis and management of thrombotic microangiopathy in Australia and New Zealand. Nephrology (Carlton) 2018;23:507–517. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
