

NMIHBA results from hypomorphic PRUNE1 variants that lack short-chain exopolyphosphatase activity
- PMID: 33105479
- PMCID: PMC7788287
- DOI: 10.1093/hmg/ddaa237
NMIHBA results from hypomorphic PRUNE1 variants that lack short-chain exopolyphosphatase activity
Abstract
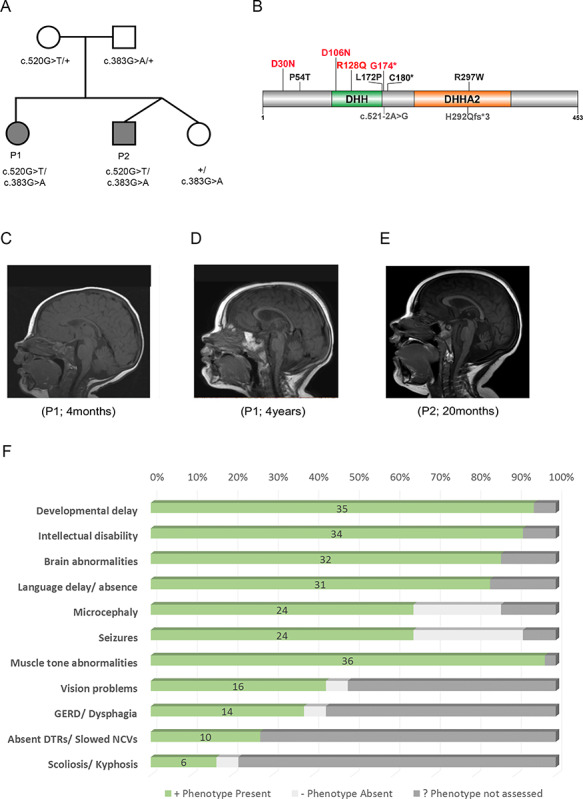
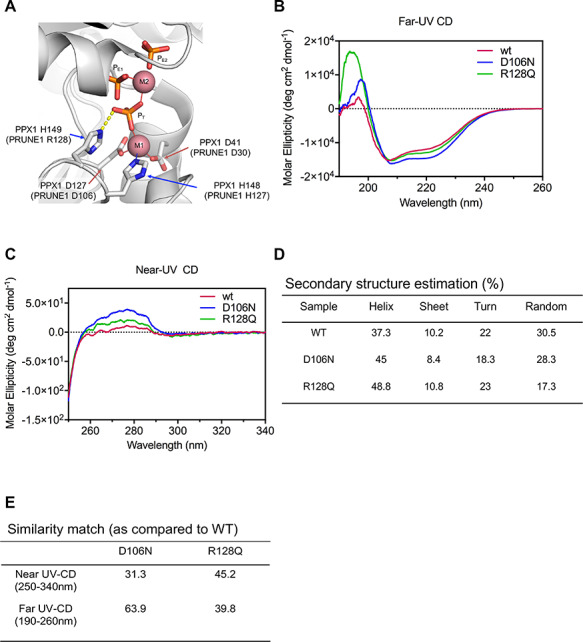
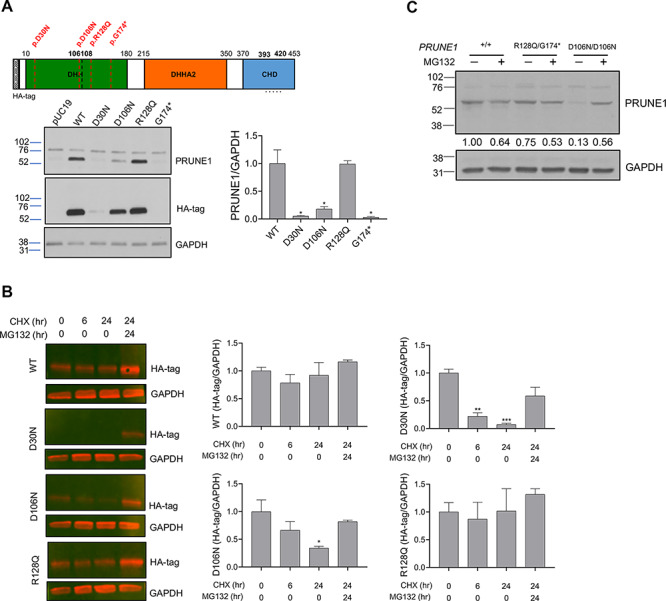
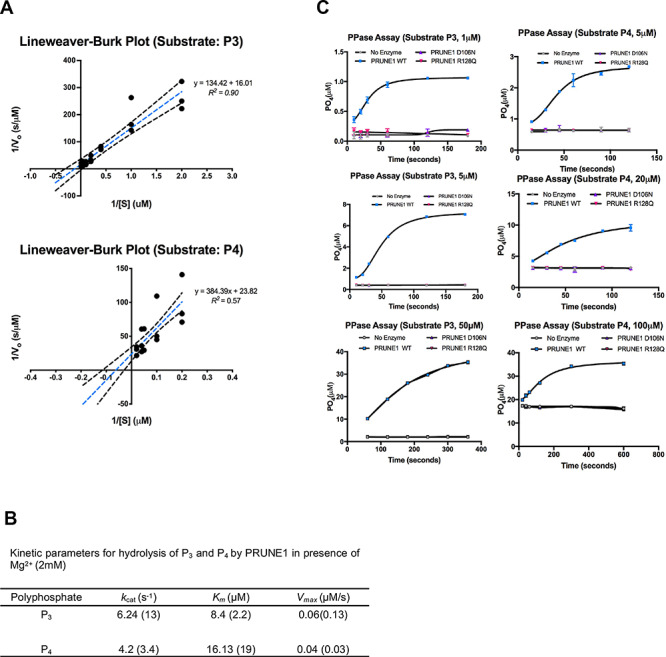
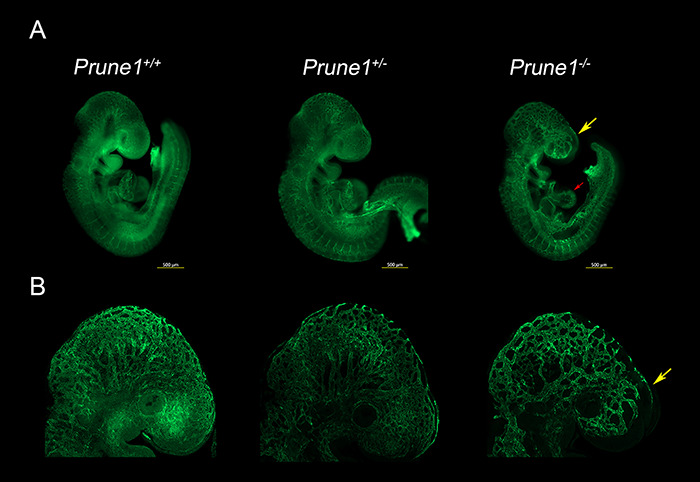
Neurodevelopmental disorder with microcephaly, hypotonia and variable brain anomalies (NMIHBA) is an autosomal recessive neurodevelopmental and neurodegenerative disorder characterized by global developmental delay and severe intellectual disability. Microcephaly, progressive cortical atrophy, cerebellar hypoplasia and delayed myelination are neurological hallmarks in affected individuals. NMIHBA is caused by biallelic variants in PRUNE1 encoding prune exopolyphosphatase 1. We provide in-depth clinical description of two affected siblings harboring compound heterozygous variant alleles, c.383G > A (p.Arg128Gln), c.520G > T (p.Gly174*) in PRUNE1. To gain insights into disease biology, we biochemically characterized missense variants within the conserved N-terminal aspartic acid-histidine-histidine (DHH) motif and provide evidence that they result in the destabilization of protein structure and/or loss of exopolyphosphatase activity. Genetic ablation of Prune1 results in midgestational lethality in mice, associated with perturbations to embryonic growth and vascular development. Our findings suggest that NMIHBA results from hypomorphic variant alleles in humans and underscore the potential key role of PRUNE1 exopolyphoshatase activity in neurodevelopment.
© The Author(s) 2020. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Shintani T., Uchiumi T., Yonezawa T., Salminen A., Baykov A.A., Lahti R. and Hachimori A. (1998) Cloning and expression of a unique inorganic pyrophosphatase from Bacillus subtilis: evidence for a new family of enzymes. FEBS Lett., 439, 263–266. - PubMed
-
- Wurst H. and Kornberg A. (1994) A soluble exopolyphosphatase of Saccharomyces cerevisiae. Purification and characterization. J. Biol. Chem., 269, 10996–11001. - PubMed
-
- Aravind L. and Koonin E.V. (1998) A novel family of predicted phosphoesterases includes drosophila prune protein and bacterial RecJ exonuclease. Trends Biochem. Sci., 23, 17–19. - PubMed
-
- Carotenuto P., Marino N., Bello A.M., D'Angelo A., Di Porzio U., Lombardi D. and Zollo M. (2006) PRUNE and NM23-M1 expression in embryonic and adult mouse brain. J. Bioenerg. Biomembr., 38, 233–246. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
