

Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches
- PMID: 33105639
- PMCID: PMC7659972
- DOI: 10.3390/ijms21217819
Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches
Abstract
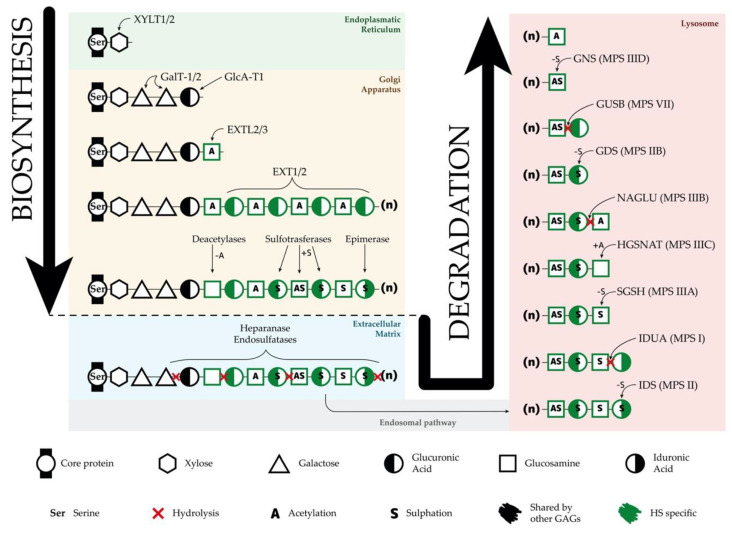
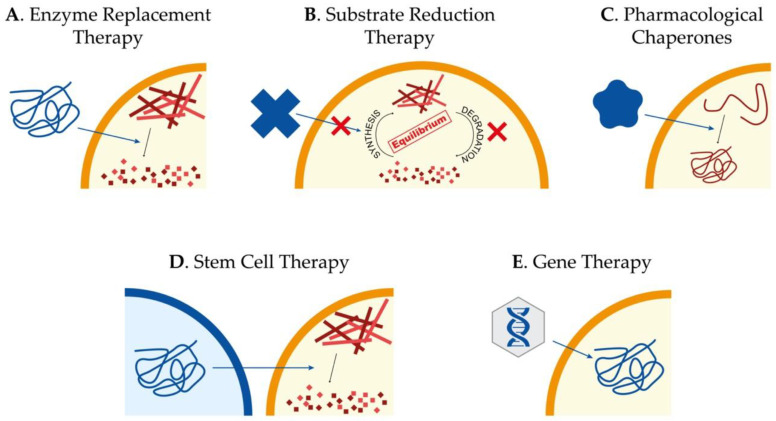
Sanfilippo syndrome or mucopolysaccharidosis III is a lysosomal storage disorder caused by mutations in genes responsible for the degradation of heparan sulfate, a glycosaminoglycan located in the extracellular membrane. Undegraded heparan sulfate molecules accumulate within lysosomes leading to cellular dysfunction and pathology in several organs, with severe central nervous system degeneration as the main phenotypical feature. The exact molecular and cellular mechanisms by which impaired degradation and storage lead to cellular dysfunction and neuronal degeneration are still not fully understood. Here, we compile the knowledge on this issue and review all available animal and cellular models that can be used to contribute to increase our understanding of Sanfilippo syndrome disease mechanisms. Moreover, we provide an update in advances regarding the different and most successful therapeutic approaches that are currently under study to treat Sanfilippo syndrome patients and discuss the potential of new tools such as induced pluripotent stem cells to be used for disease modeling and therapy development.
Keywords: Sanfilippo syndrome; animal models; cellular models; heparan sulfate; induced pluripotent stem cells; lysosomal storage disorders; mucopolysaccharidosis III; therapeutic approaches.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Neufeld E.F., Muenzer J. The Mucopolysaccharidoses. In: Scriver C.R., Valle D.L., Antonarakis S., Ballabio A., Beaudet A.L., Mitchell G.A., editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York, NY, USA: 2001. pp. 3421–3452. - DOI
-
- Sanfilippo S.J., Good R.A., Podosin R., Langer L. Mental Retardation Associated with Acid Mucopolysacchariduria (Heparitin Sulfate Type) J. Pediatr-Us. 1963;63:837–838. doi: 10.1016/S0022-3476(63)80279-6. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
