

RNA-binding protein DDX3 mediates posttranscriptional regulation of androgen receptor: A mechanism of castration resistance
- PMID: 33106406
- PMCID: PMC7668093
- DOI: 10.1073/pnas.2008479117
RNA-binding protein DDX3 mediates posttranscriptional regulation of androgen receptor: A mechanism of castration resistance
Abstract
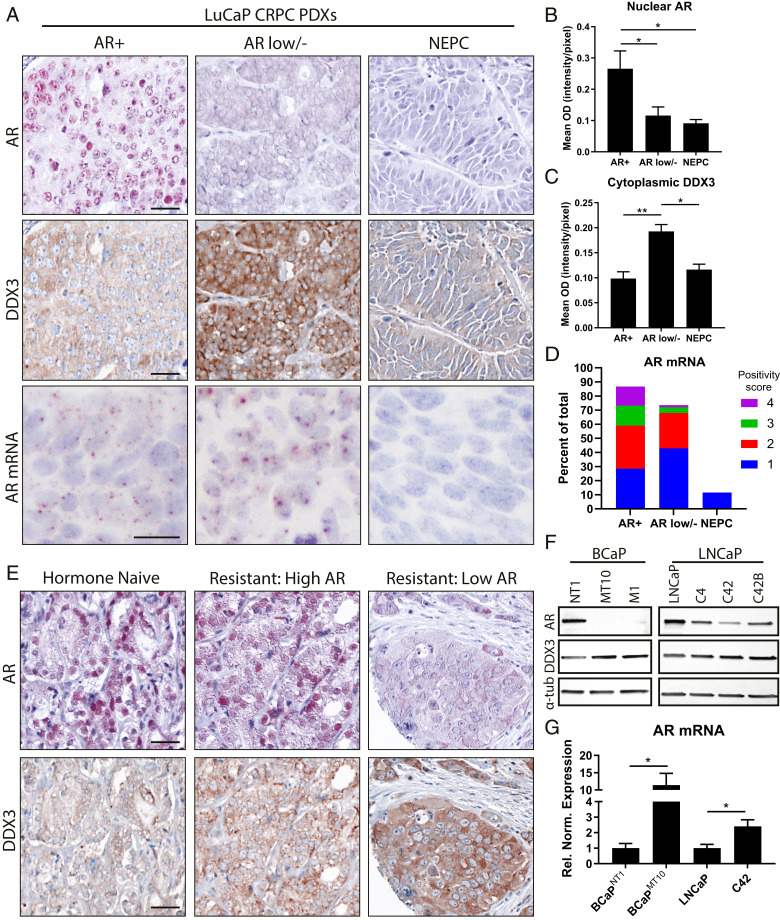
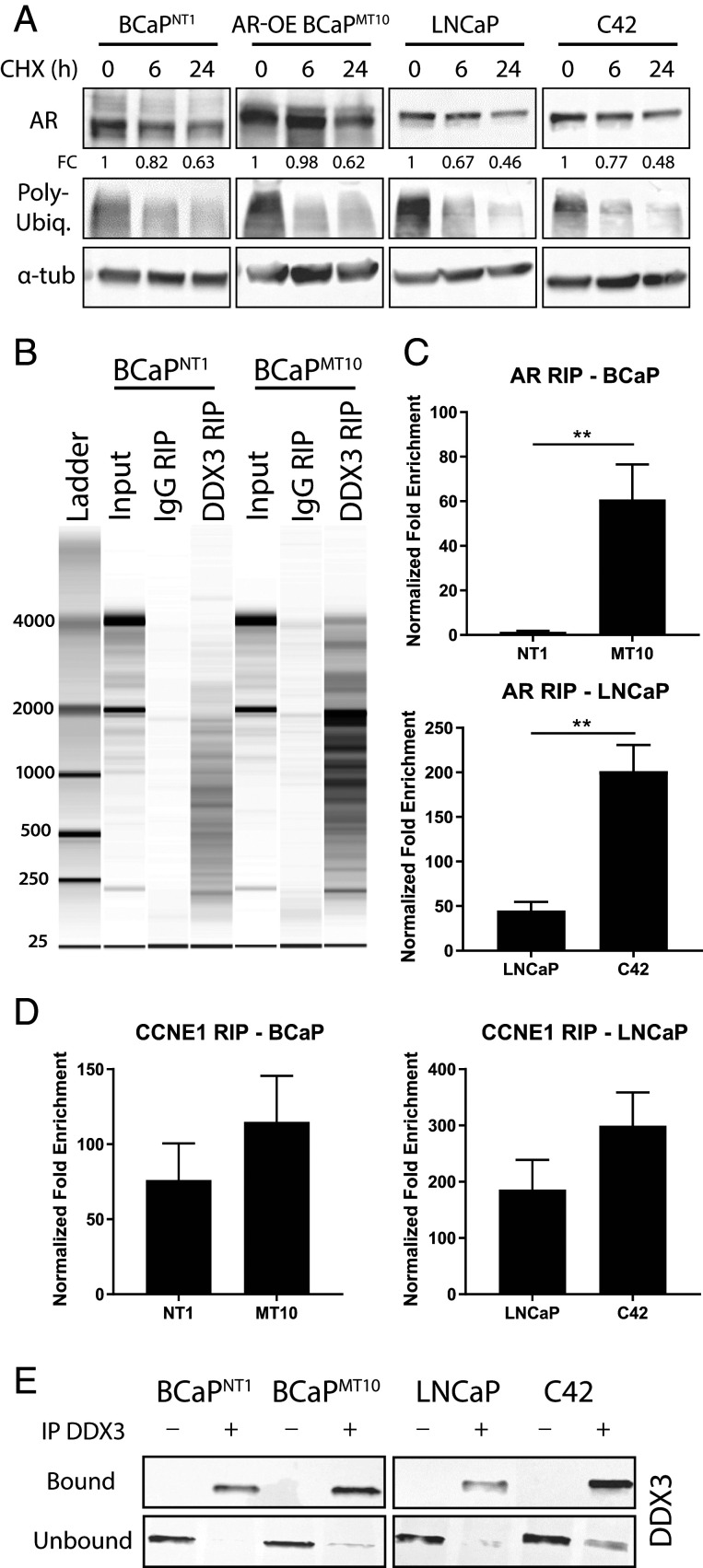
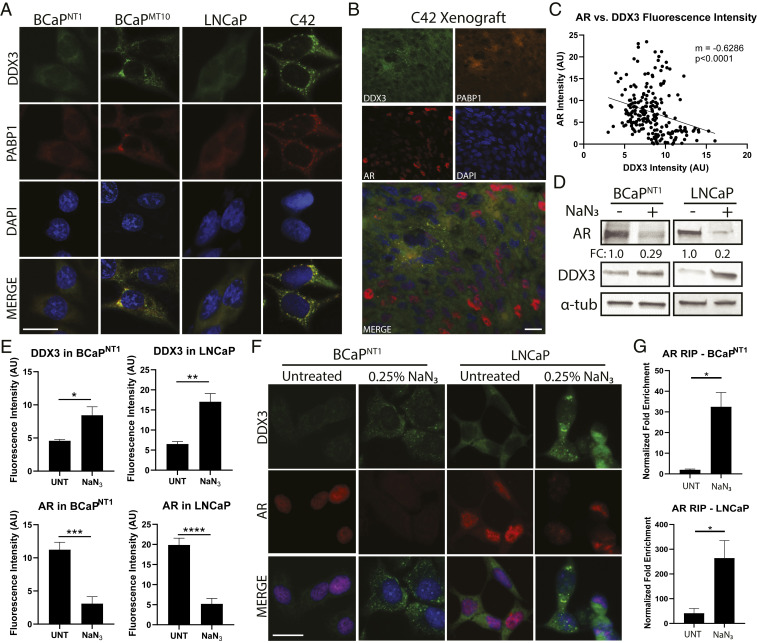
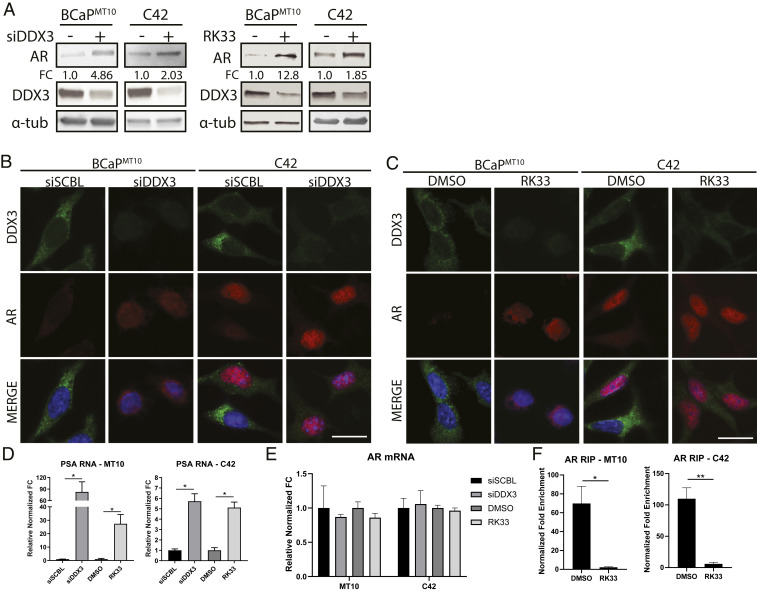
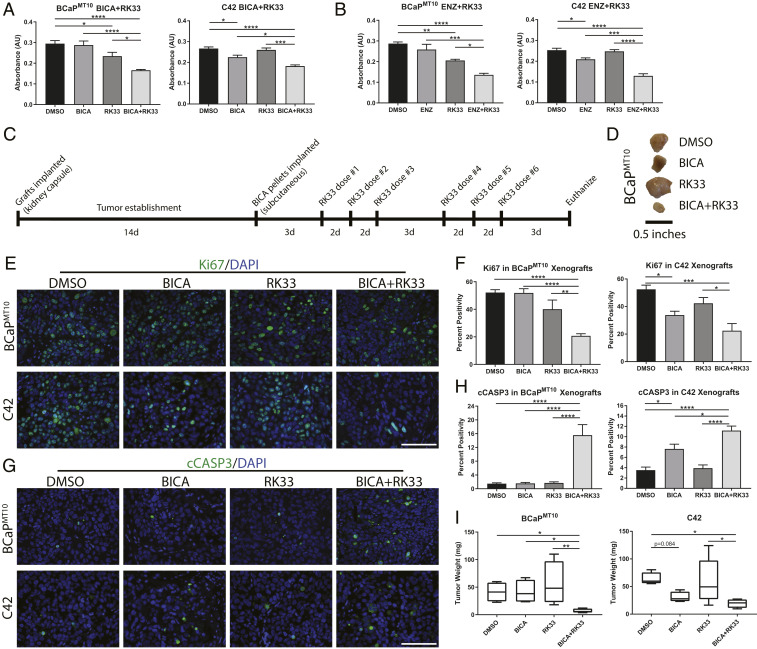
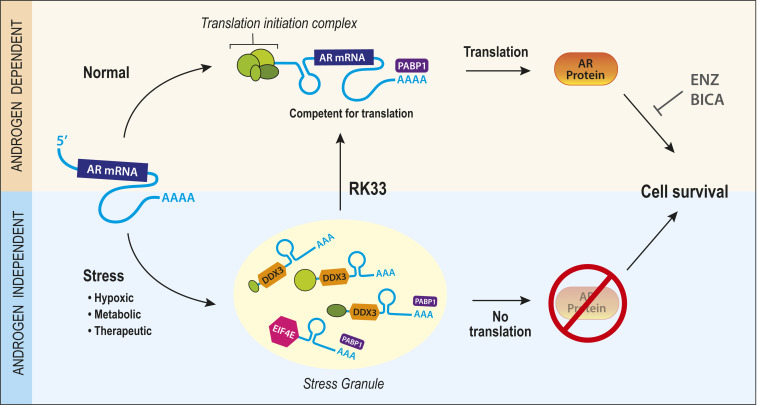
Prostate cancer (CaP) driven by androgen receptor (AR) is treated with androgen deprivation; however, therapy failure results in lethal castration-resistant prostate cancer (CRPC). AR-low/negative (ARL/-) CRPC subtypes have recently been characterized and cannot be targeted by hormonal therapies, resulting in poor prognosis. RNA-binding protein (RBP)/helicase DDX3 (DEAD-box helicase 3 X-linked) is a key component of stress granules (SG) and is postulated to affect protein translation. Here, we investigated DDX3-mediated posttranscriptional regulation of AR mRNA (messenger RNA) in CRPC. Using patient samples and preclinical models, we objectively quantified DDX3 and AR expression in ARL/- CRPC. We utilized CRPC models to identify DDX3:AR mRNA complexes by RNA immunoprecipitation, assess the effects of DDX3 gain/loss-of-function on AR expression and signaling, and address clinical implications of targeting DDX3 by assessing sensitivity to AR-signaling inhibitors (ARSI) in CRPC xenografts in vivo. ARL/- CRPC expressed abundant AR mRNA despite diminished levels of AR protein. DDX3 protein was highly expressed in ARL/- CRPC, where it bound to AR mRNA. Consistent with a repressive regulatory role, DDX3 localized to cytoplasmic puncta with SG marker PABP1 in CRPC. While induction of DDX3-nucleated SGs resulted in decreased AR protein expression, inhibiting DDX3 was sufficient to restore 1) AR protein expression, 2) AR signaling, and 3) sensitivity to ARSI in vitro and in vivo. Our findings implicate the RBP protein DDX3 as a mechanism of posttranscriptional regulation for AR in CRPC. Clinically, DDX3 may be targetable for sensitizing ARL/- CRPC to AR-directed therapies.
Keywords: androgen independence; castration-resistant prostate cancer; double-negative prostate cancer; posttranscriptional regulation; prostate cancer.
Conflict of interest statement
The authors declare no competing interest.
Figures
Comment in
-
Uro-Science.J Urol. 2021 Aug;206(2):480-482. doi: 10.1097/JU.0000000000001849. Epub 2021 May 12. J Urol. 2021. PMID: 33975458 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
