

Characterization of systemic genomic instability in budding yeast
- PMID: 33106418
- PMCID: PMC7668020
- DOI: 10.1073/pnas.2010303117
Characterization of systemic genomic instability in budding yeast
Abstract
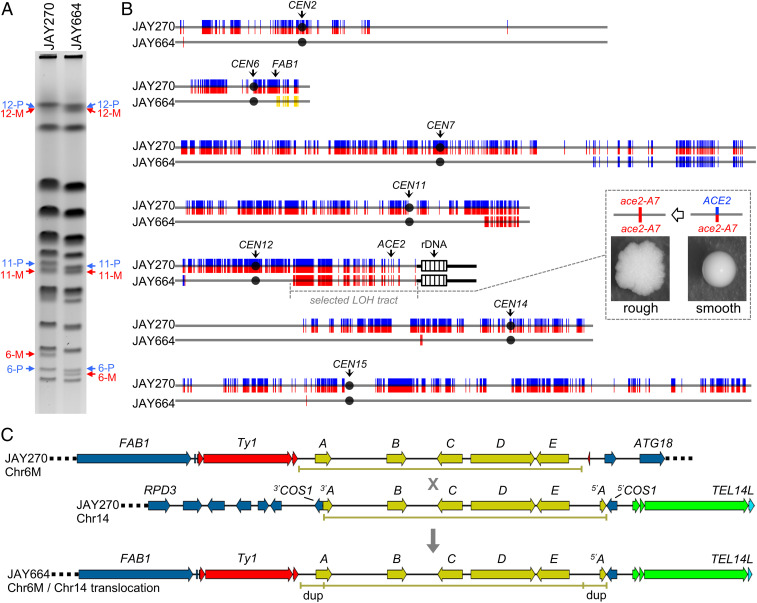
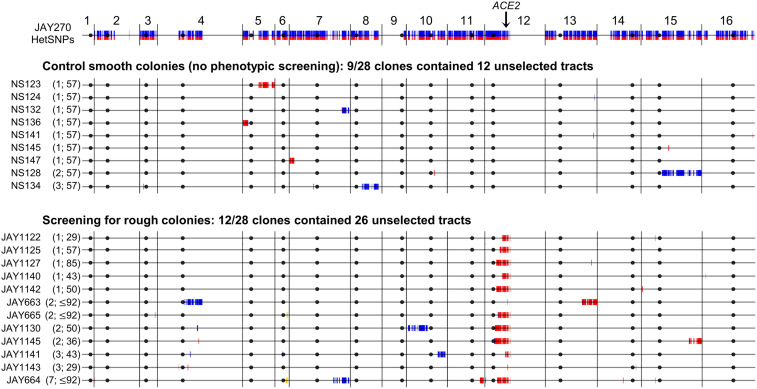
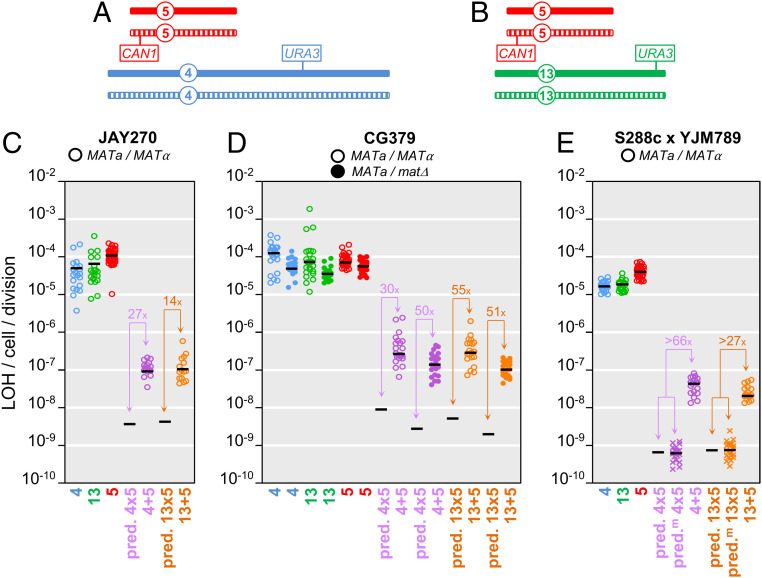
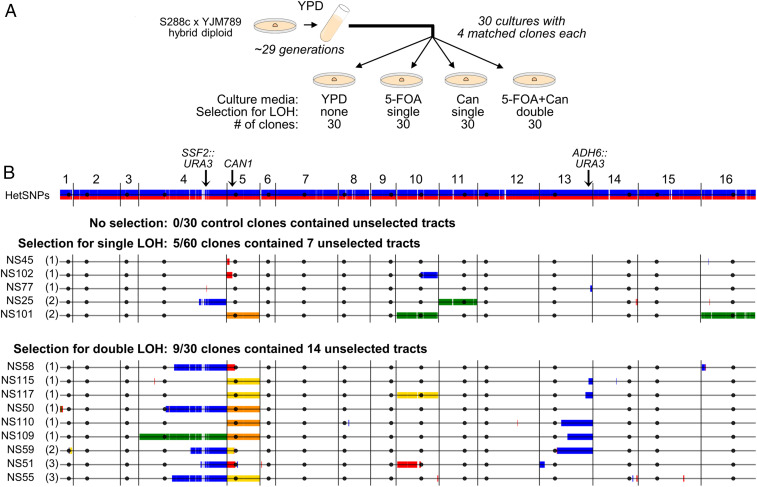
Conventional models of genome evolution are centered around the principle that mutations form independently of each other and build up slowly over time. We characterized the occurrence of bursts of genome-wide loss-of-heterozygosity (LOH) in Saccharomyces cerevisiae, providing support for an additional nonindependent and faster mode of mutation accumulation. We initially characterized a yeast clone isolated for carrying an LOH event at a specific chromosome site, and surprisingly found that it also carried multiple unselected rearrangements elsewhere in its genome. Whole-genome analysis of over 100 additional clones selected for carrying primary LOH tracts revealed that they too contained unselected structural alterations more often than control clones obtained without any selection. We also measured the rates of coincident LOH at two different chromosomes and found that double LOH formed at rates 14- to 150-fold higher than expected if the two underlying single LOH events occurred independently of each other. These results were consistent across different strain backgrounds and in mutants incapable of entering meiosis. Our results indicate that a subset of mitotic cells within a population can experience discrete episodes of systemic genomic instability, when the entire genome becomes vulnerable and multiple chromosomal alterations can form over a narrow time window. They are reminiscent of early reports from the classic yeast genetics literature, as well as recent studies in humans, both in cancer and genomic disorder contexts. The experimental model we describe provides a system to further dissect the fundamental biological processes responsible for punctuated bursts of structural genomic variation.
Keywords: genomic instability; loss-of-heterozygosity; mitotic recombination.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
