

Comprehensive characterization of amino acid positions in protein structures reveals molecular effect of missense variants
- PMID: 33106425
- PMCID: PMC7668189
- DOI: 10.1073/pnas.2002660117
Comprehensive characterization of amino acid positions in protein structures reveals molecular effect of missense variants
Abstract
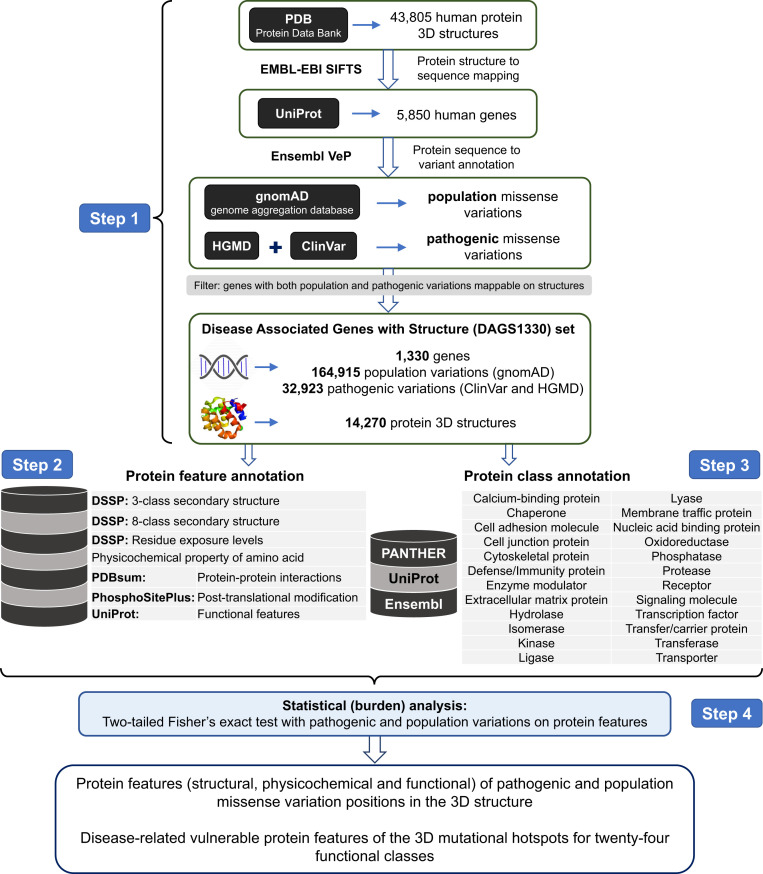
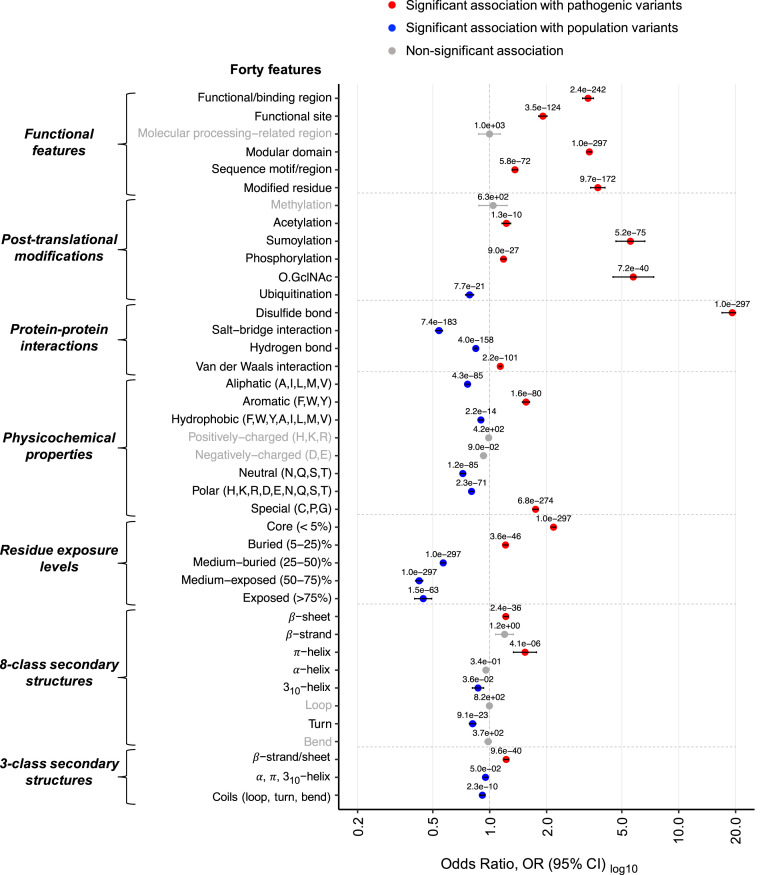
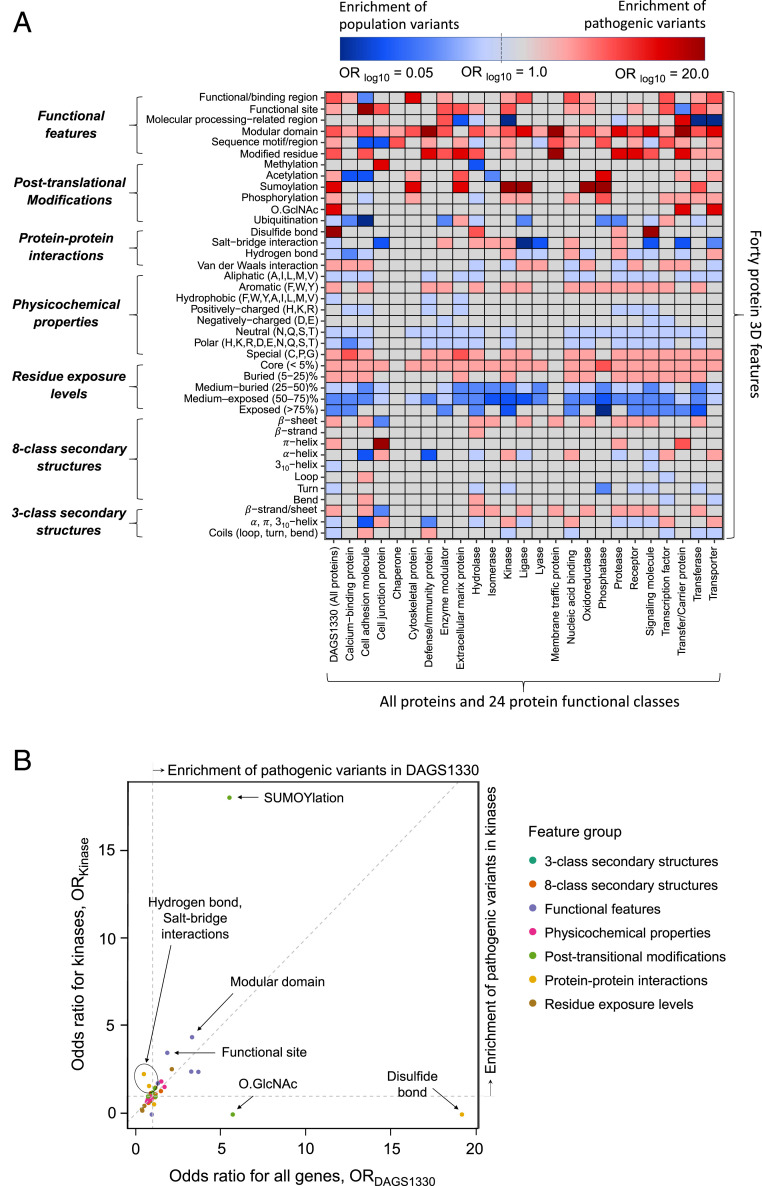
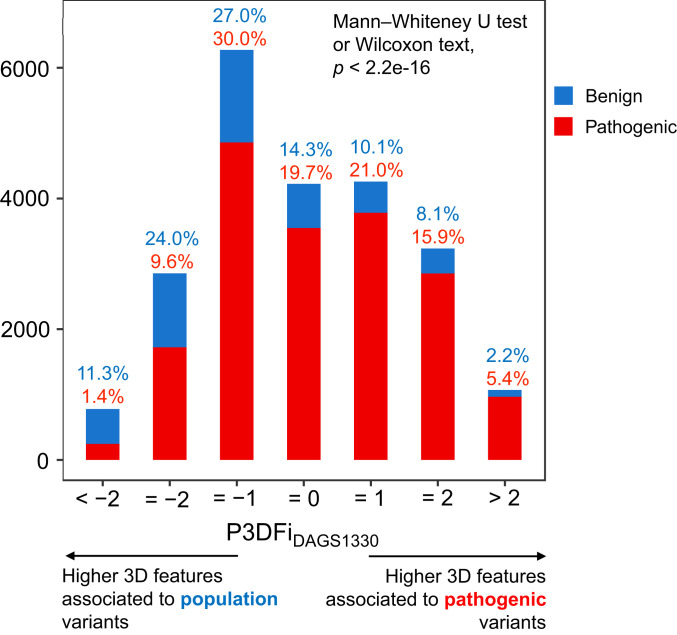
Interpretation of the colossal number of genetic variants identified from sequencing applications is one of the major bottlenecks in clinical genetics, with the inference of the effect of amino acid-substituting missense variations on protein structure and function being especially challenging. Here we characterize the three-dimensional (3D) amino acid positions affected in pathogenic and population variants from 1,330 disease-associated genes using over 14,000 experimentally solved human protein structures. By measuring the statistical burden of variations (i.e., point mutations) from all genes on 40 3D protein features, accounting for the structural, chemical, and functional context of the variations' positions, we identify features that are generally associated with pathogenic and population missense variants. We then perform the same amino acid-level analysis individually for 24 protein functional classes, which reveals unique characteristics of the positions of the altered amino acids: We observe up to 46% divergence of the class-specific features from the general characteristics obtained by the analysis on all genes, which is consistent with the structural diversity of essential regions across different protein classes. We demonstrate that the function-specific 3D features of the variants match the readouts of mutagenesis experiments for BRCA1 and PTEN, and positively correlate with an independent set of clinically interpreted pathogenic and benign missense variants. Finally, we make our results available through a web server to foster accessibility and downstream research. Our findings represent a crucial step toward translational genetics, from highlighting the impact of mutations on protein structure to rationalizing the variants' pathogenicity in terms of the perturbed molecular mechanisms.
Keywords: 3D mutational hotspot; disease variation effect; machine learning; missense variant interpretation; protein structure and function.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
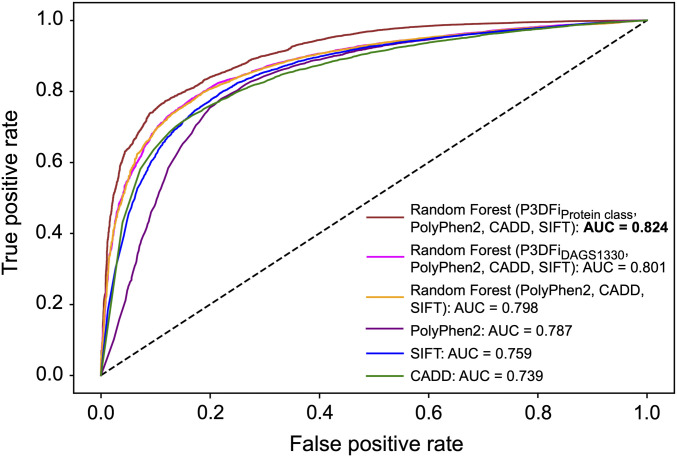
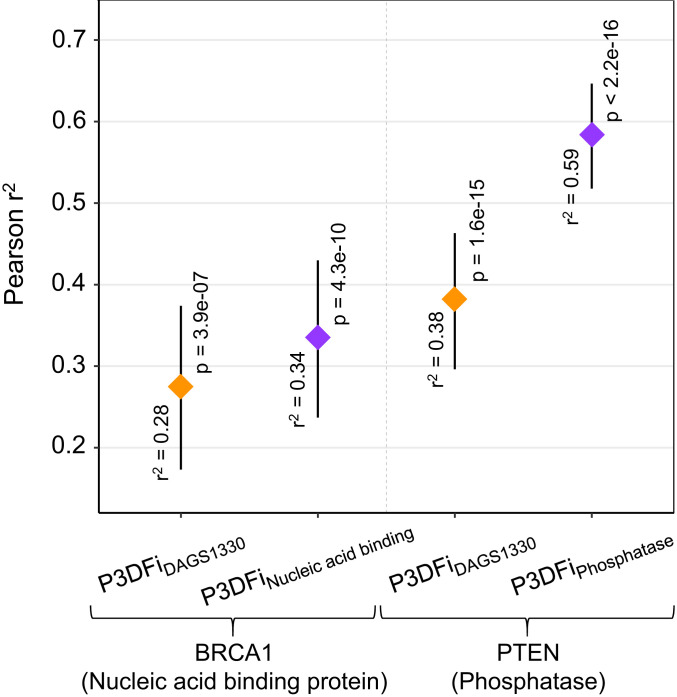

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
