

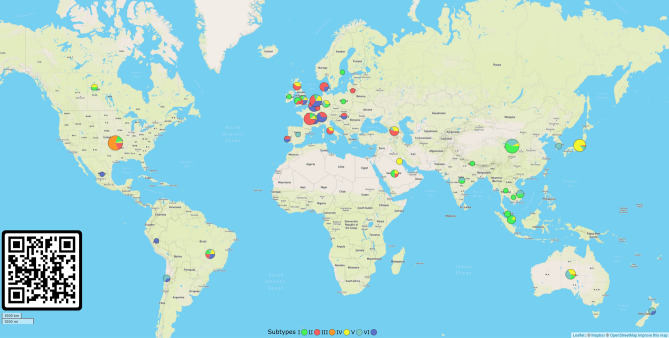
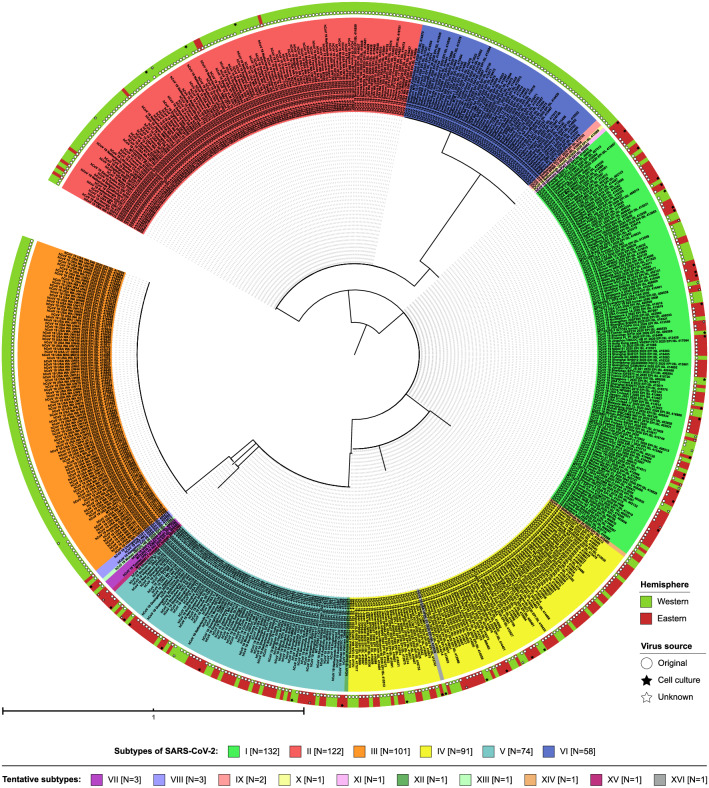
The global population of SARS-CoV-2 is composed of six major subtypes
- PMID: 33106569
- PMCID: PMC7588421
- DOI: 10.1038/s41598-020-74050-8
The global population of SARS-CoV-2 is composed of six major subtypes
Abstract
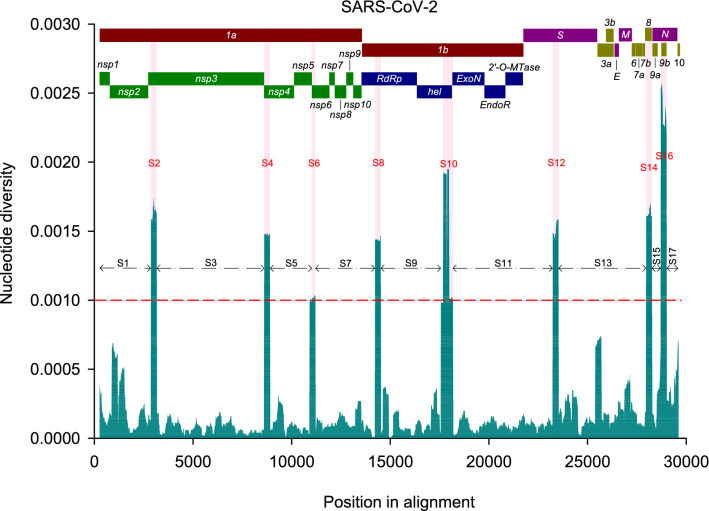
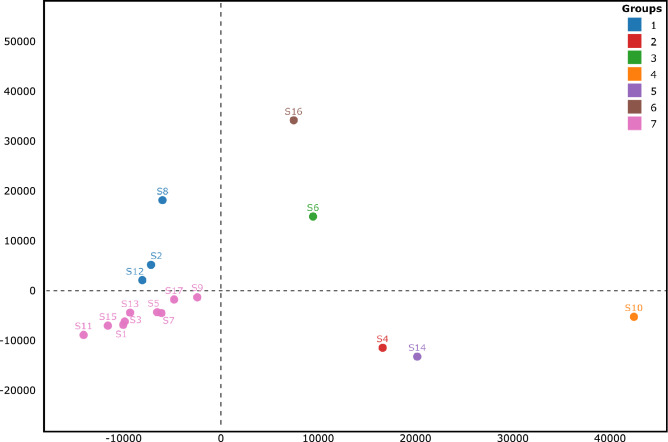
The World Health Organization characterized COVID-19 as a pandemic in March 2020, the second pandemic of the twenty-first century. Expanding virus populations, such as that of SARS-CoV-2, accumulate a number of narrowly shared polymorphisms, imposing a confounding effect on traditional clustering methods. In this context, approaches that reduce the complexity of the sequence space occupied by the SARS-CoV-2 population are necessary for robust clustering. Here, we propose subdividing the global SARS-CoV-2 population into six well-defined subtypes and 10 poorly represented genotypes named tentative subtypes by focusing on the widely shared polymorphisms in nonstructural (nsp3, nsp4, nsp6, nsp12, nsp13 and nsp14) cistrons and structural (spike and nucleocapsid) and accessory (ORF8) genes. The six subtypes and the additional genotypes showed amino acid replacements that might have phenotypic implications. Notably, three mutations (one of them in the Spike protein) were responsible for the geographical segregation of subtypes. We hypothesize that the virus subtypes detected in this study are records of the early stages of SARS-CoV-2 diversification that were randomly sampled to compose the virus populations around the world. The genetic structure determined for the SARS-CoV-2 population provides substantial guidelines for maximizing the effectiveness of trials for testing candidate vaccines or drugs.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- WHO. WHO Director-General’s remarks at the media briefing on 2019-nCoV on 11 February 2020. WHO website.https://www.who.int/dg/speeches/detail/who-directo (2020). Accessed 10 Apr 2020.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
