

Timing the origin of eukaryotic cellular complexity with ancient duplications
- PMID: 33106602
- PMCID: PMC7610411
- DOI: 10.1038/s41559-020-01320-z
Timing the origin of eukaryotic cellular complexity with ancient duplications
Abstract
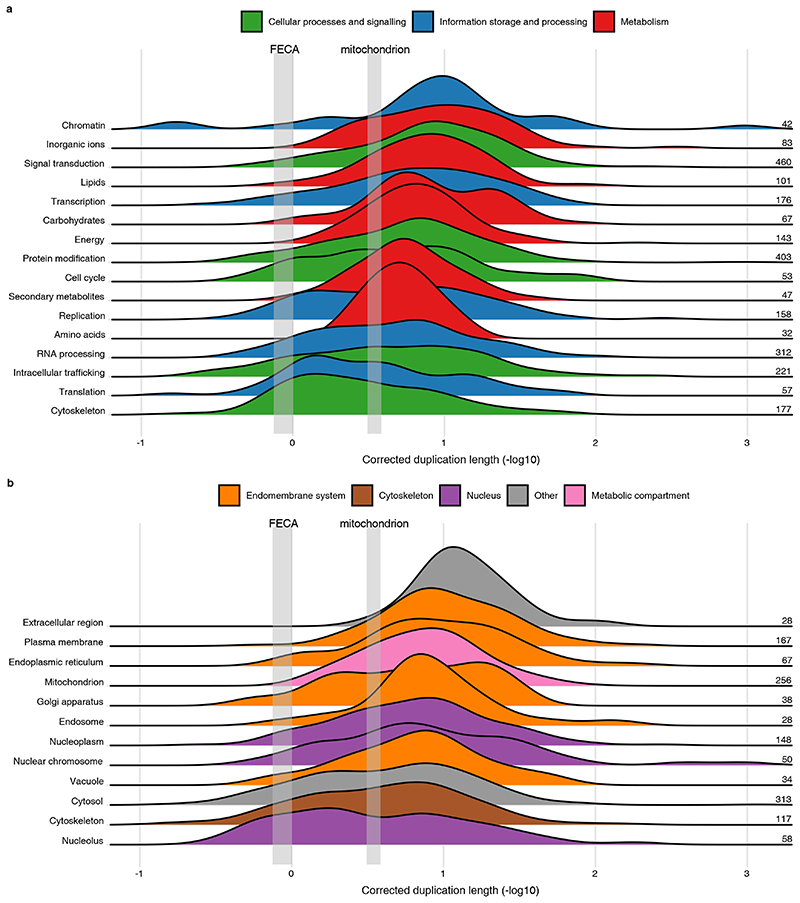
Eukaryogenesis is one of the most enigmatic evolutionary transitions, during which simple prokaryotic cells gave rise to complex eukaryotic cells. While evolutionary intermediates are lacking, gene duplications provide information on the order of events by which eukaryotes originated. Here we use a phylogenomics approach to reconstruct successive steps during eukaryogenesis. We find that gene duplications roughly doubled the proto-eukaryotic gene repertoire, with families inherited from the Asgard archaea-related host being duplicated most. By relatively timing events using phylogenetic distances, we inferred that duplications in cytoskeletal and membrane-trafficking families were among the earliest events, whereas most other families expanded predominantly after mitochondrial endosymbiosis. Altogether, we infer that the host that engulfed the proto-mitochondrion had some eukaryote-like complexity, which drastically increased upon mitochondrial acquisition. This scenario bridges the signs of complexity observed in Asgard archaeal genomes to the proposed role of mitochondria in triggering eukaryogenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures









References
-
- Dacks JB, et al. The changing view of eukaryogenesis – fossils, cells, lineages and how they all come together. J Cell Sci. 2016;129:3695–3703. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
