

Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer's and Parkinson's diseases
- PMID: 33106633
- PMCID: PMC7606627
- DOI: 10.1038/s41588-020-00721-x
Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer's and Parkinson's diseases
Abstract
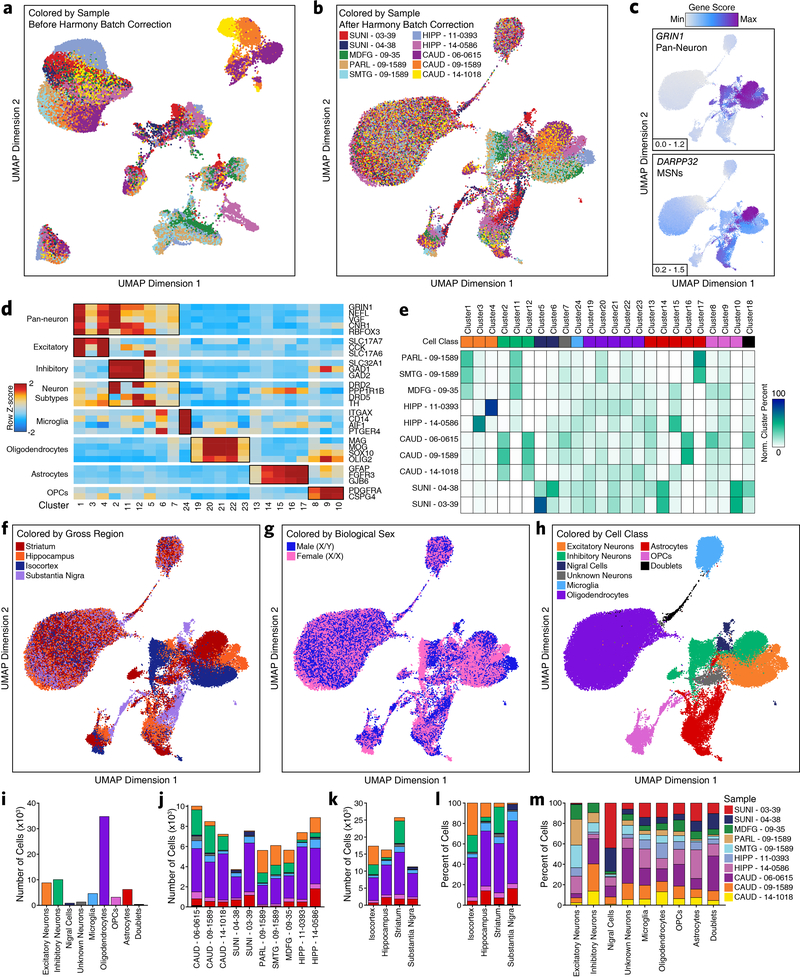
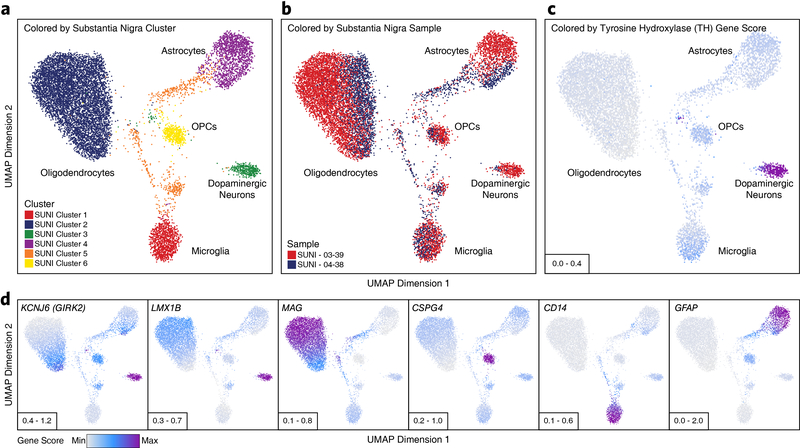
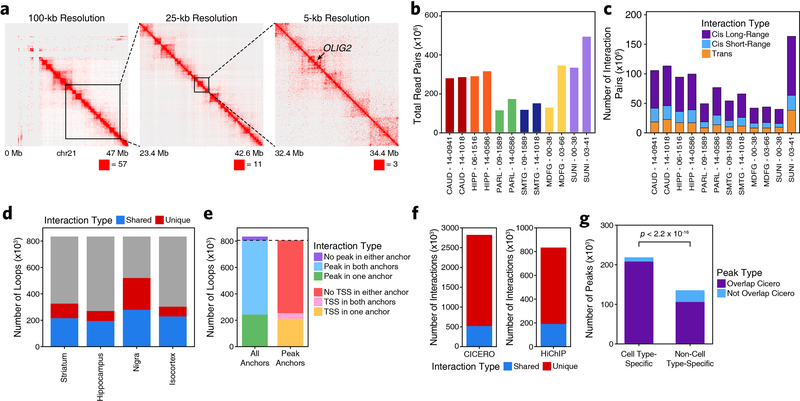
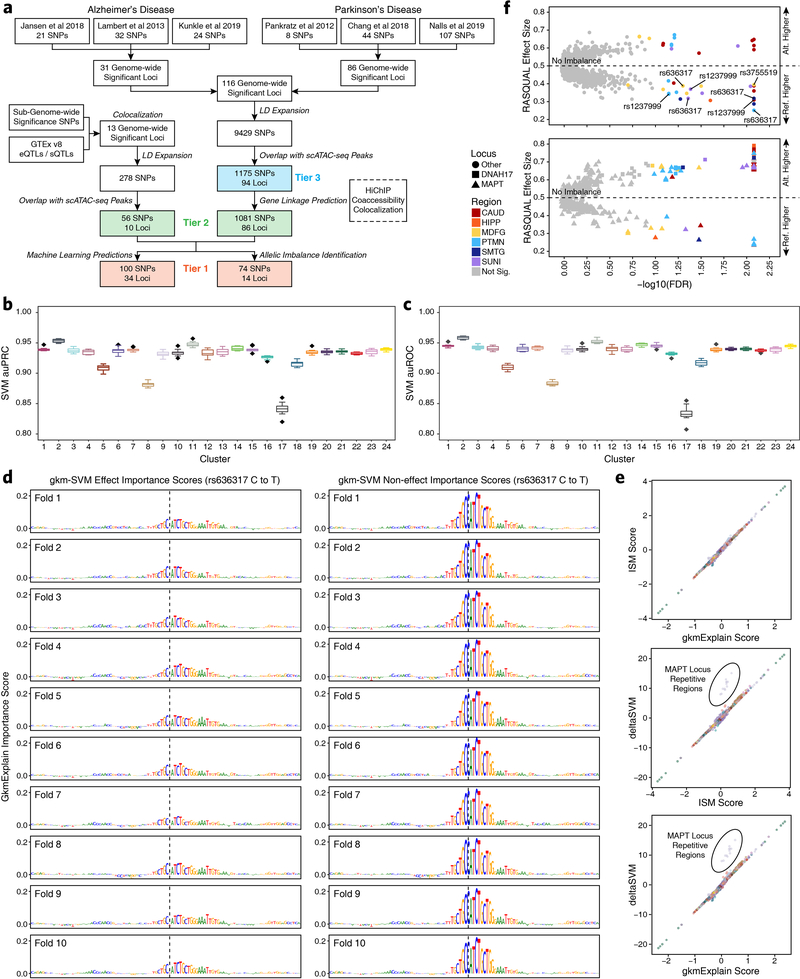
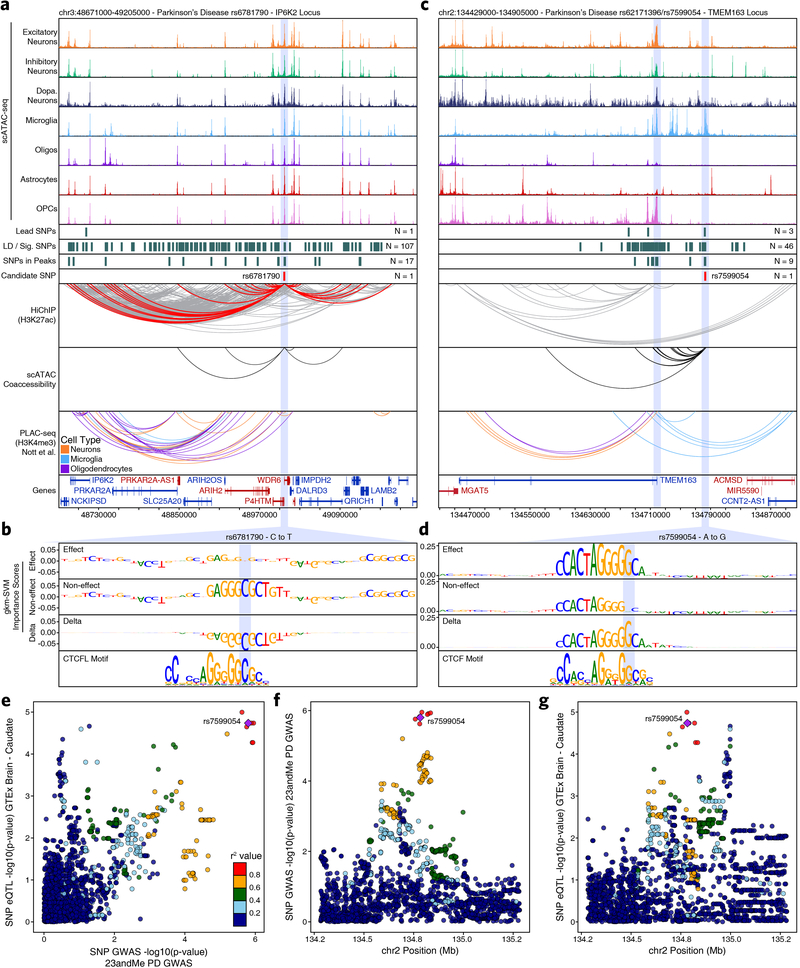
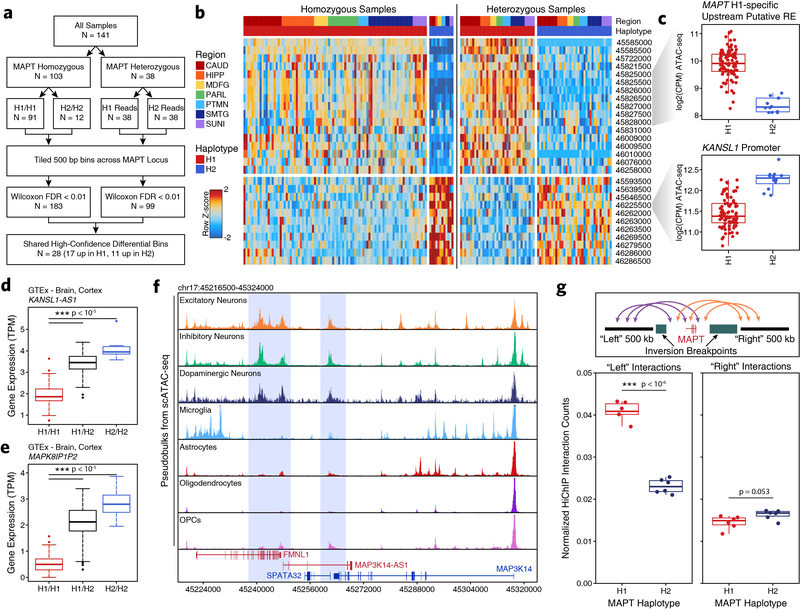
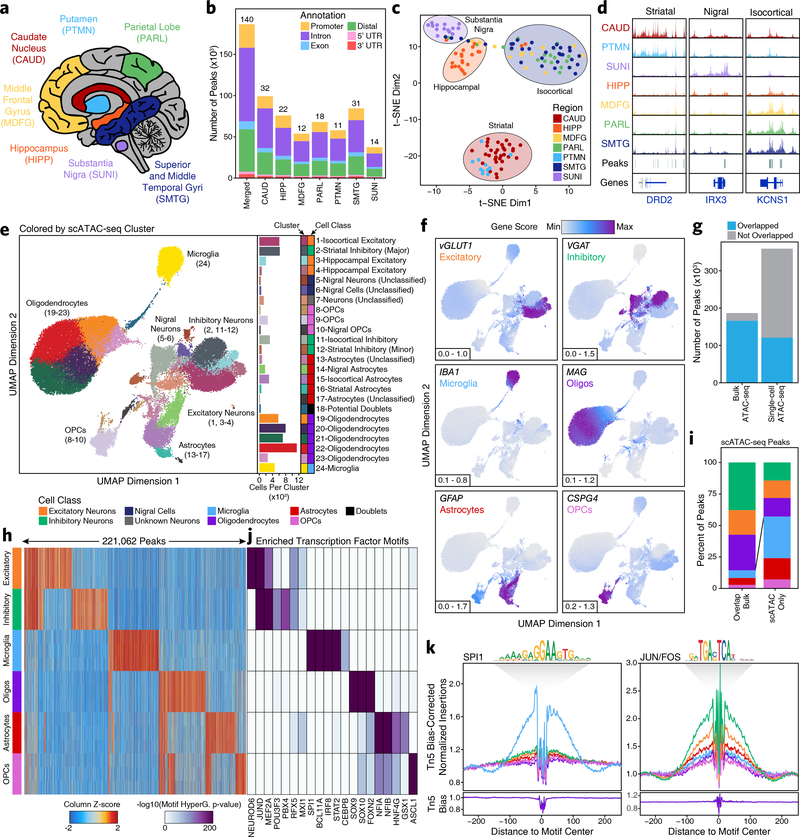
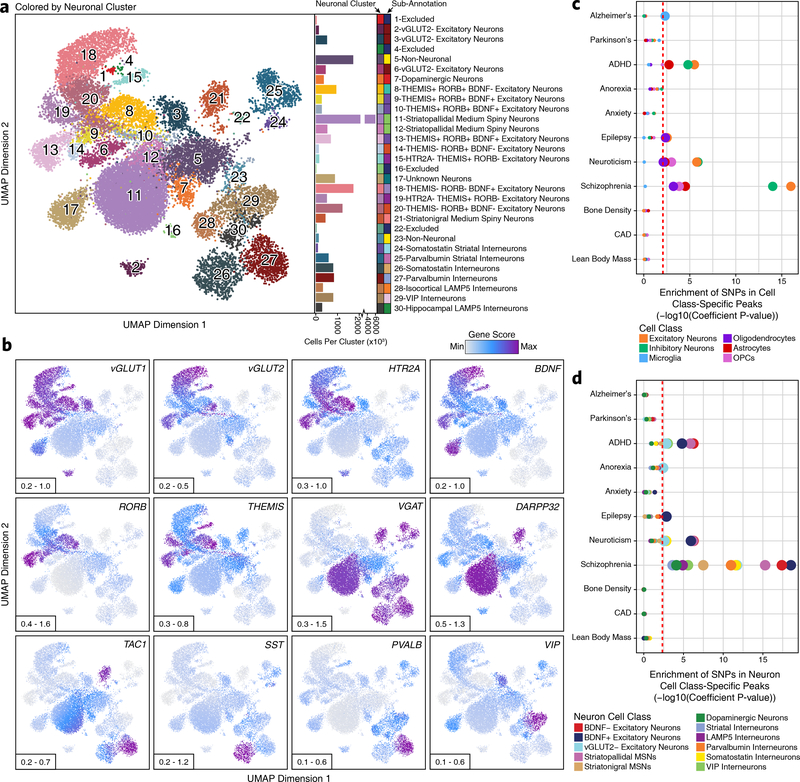
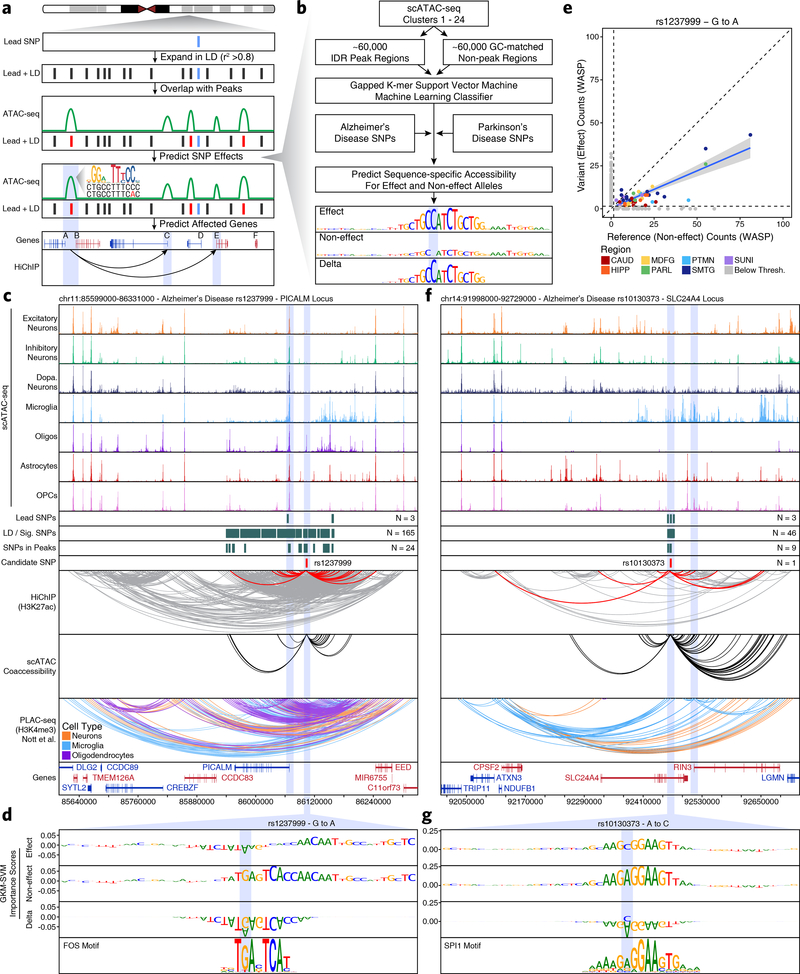
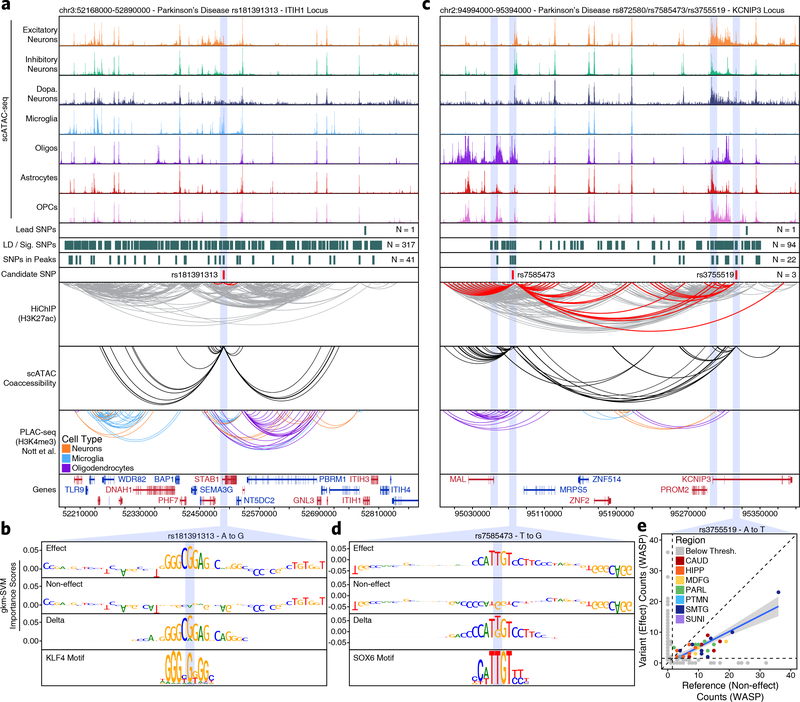
Genome-wide association studies of neurological diseases have identified thousands of variants associated with disease phenotypes. However, most of these variants do not alter coding sequences, making it difficult to assign their function. Here, we present a multi-omic epigenetic atlas of the adult human brain through profiling of single-cell chromatin accessibility landscapes and three-dimensional chromatin interactions of diverse adult brain regions across a cohort of cognitively healthy individuals. We developed a machine-learning classifier to integrate this multi-omic framework and predict dozens of functional SNPs for Alzheimer's and Parkinson's diseases, nominating target genes and cell types for previously orphaned loci from genome-wide association studies. Moreover, we dissected the complex inverted haplotype of the MAPT (encoding tau) Parkinson's disease risk locus, identifying putative ectopic regulatory interactions in neurons that may mediate this disease association. This work expands understanding of inherited variation and provides a roadmap for the epigenomic dissection of causal regulatory variation in disease.
Conflict of interest statement
COMPETING INTERESTS STATEMENT
H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, and an advisor to 10x Genomics, Arsenal Biosciences, Spring Discovery. S.B.M. is on the scientific advisory board of MyOme. A.K. is a consultant for Biogen Inc. A.S. is a consultant for MyoKardia. W.J.G. is a consultant for Guardant Health, 10x Genomics, and Protillion Biosciences.
Figures




Comment in
-
Applying Artificial Intelligence to Multi-Omic Data: New Functional Variants in Parkinson's Disease.Mov Disord. 2021 Feb;36(2):347. doi: 10.1002/mds.28481. Epub 2021 Jan 17. Mov Disord. 2021. PMID: 33458867 No abstract available.
References
REFERENCES (MAIN TEXT)
METHODS-ONLY REFERENCES
-
- Li Z et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 49, 1576–1583 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- R00 AG059918/AG/NIA NIH HHS/United States
- U01 HG009431/HG/NHGRI NIH HHS/United States
- P30 AG066515/AG/NIA NIH HHS/United States
- K99 AG059918/AG/NIA NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- P50 AG005136/AG/NIA NIH HHS/United States
- UF1 AG057707/AG/NIA NIH HHS/United States
- P50 HG007735/HG/NHGRI NIH HHS/United States
- T15 LM007033/LM/NLM NIH HHS/United States
- P30 AG019610/AG/NIA NIH HHS/United States
- S10 OD018220/OD/NIH HHS/United States
- RF1 AG053959/AG/NIA NIH HHS/United States
- S10 OD025212/OD/NIH HHS/United States
- P50 NS062684/NS/NINDS NIH HHS/United States
- P30 AG066509/AG/NIA NIH HHS/United States
- P50 AG047366/AG/NIA NIH HHS/United States
- R01 AG066490/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
