

Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease
- PMID: 33108101
- PMCID: PMC7847551
- DOI: 10.1056/NEJMoa2026834
Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease
Abstract
Background: Adult-onset inflammatory syndromes often manifest with overlapping clinical features. Variants in ubiquitin-related genes, previously implicated in autoinflammatory disease, may define new disorders.
Methods: We analyzed peripheral-blood exome sequence data independent of clinical phenotype and inheritance pattern to identify deleterious mutations in ubiquitin-related genes. Sanger sequencing, immunoblotting, immunohistochemical testing, flow cytometry, and transcriptome and cytokine profiling were performed. CRISPR-Cas9-edited zebrafish were used as an in vivo model to assess gene function.
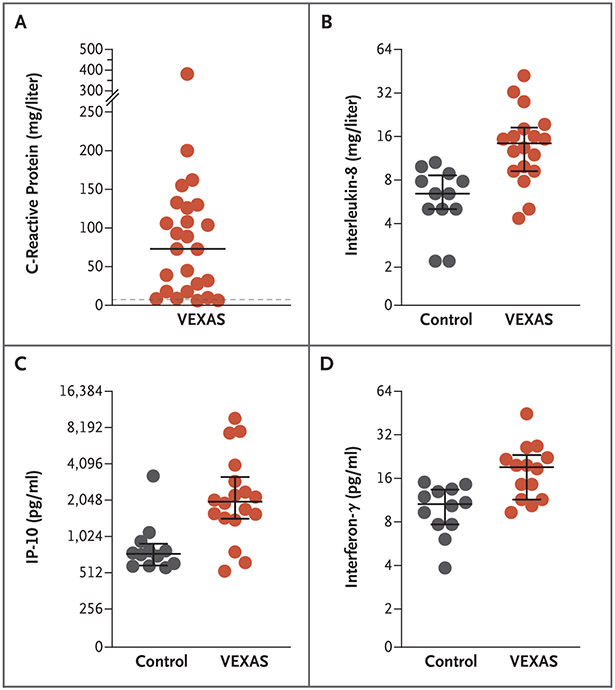
Results: We identified 25 men with somatic mutations affecting methionine-41 (p.Met41) in UBA1, the major E1 enzyme that initiates ubiquitylation. (The gene UBA1 lies on the X chromosome.) In such patients, an often fatal, treatment-refractory inflammatory syndrome develops in late adulthood, with fevers, cytopenias, characteristic vacuoles in myeloid and erythroid precursor cells, dysplastic bone marrow, neutrophilic cutaneous and pulmonary inflammation, chondritis, and vasculitis. Most of these 25 patients met clinical criteria for an inflammatory syndrome (relapsing polychondritis, Sweet's syndrome, polyarteritis nodosa, or giant-cell arteritis) or a hematologic condition (myelodysplastic syndrome or multiple myeloma) or both. Mutations were found in more than half the hematopoietic stem cells, including peripheral-blood myeloid cells but not lymphocytes or fibroblasts. Mutations affecting p.Met41 resulted in loss of the canonical cytoplasmic isoform of UBA1 and in expression of a novel, catalytically impaired isoform initiated at p.Met67. Mutant peripheral-blood cells showed decreased ubiquitylation and activated innate immune pathways. Knockout of the cytoplasmic UBA1 isoform homologue in zebrafish caused systemic inflammation.
Conclusions: Using a genotype-driven approach, we identified a disorder that connects seemingly unrelated adult-onset inflammatory syndromes. We named this disorder the VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome. (Funded by the NIH Intramural Research Programs and the EU Horizon 2020 Research and Innovation Program.).
Copyright © 2020 Massachusetts Medical Society.
Figures



Comment in
-
Hiding in Plain Sight - Somatic Mutation in Human Disease.N Engl J Med. 2020 Dec 31;383(27):2680-2682. doi: 10.1056/NEJMe2030754. Epub 2020 Oct 27. N Engl J Med. 2020. PMID: 33108100 No abstract available.
-
Somatic mutations cause VEXAS syndrome.Nat Rev Rheumatol. 2021 Jan;17(1):1. doi: 10.1038/s41584-020-00559-x. Nat Rev Rheumatol. 2021. PMID: 33262468 No abstract available.
-
Novel somatic mutations in UBA1 as a cause of VEXAS syndrome.Blood. 2021 Jul 1;137(26):3676-3681. doi: 10.1182/blood.2020010286. Blood. 2021. PMID: 33690815 Free PMC article.
-
Mutant UBA1 and Severe Adult-Onset Autoinflammatory Disease.N Engl J Med. 2021 Jun 3;384(22):2163. doi: 10.1056/NEJMc2102124. N Engl J Med. 2021. PMID: 34077651 No abstract available.
-
Mutant UBA1 and Severe Adult-Onset Autoinflammatory Disease.N Engl J Med. 2021 Jun 3;384(22):2163-2164. doi: 10.1056/NEJMc2102124. N Engl J Med. 2021. PMID: 34077652 No abstract available.
-
Mutant UBA1 and Severe Adult-Onset Autoinflammatory Disease.N Engl J Med. 2021 Jun 3;384(22):2164. doi: 10.1056/NEJMc2102124. N Engl J Med. 2021. PMID: 34077653 No abstract available.
-
VEXAS syndrome with systemic lupus erythematosus: expanding the spectrum of associated conditions.Arthritis Rheumatol. 2022 Feb;74(2):369-371. doi: 10.1002/art.41957. Epub 2021 Dec 27. Arthritis Rheumatol. 2022. PMID: 34463053 Free PMC article. No abstract available.
-
Hematopoietic cells vacuolation, not always a reactive event. The VEXAS syndrome.Int J Lab Hematol. 2023 Feb;45(1):e15-e16. doi: 10.1111/ijlh.13955. Epub 2022 Sep 5. Int J Lab Hematol. 2023. PMID: 36065051 No abstract available.
References
-
- Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 2017;18:832–42. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous