

An Information-Theory-Based Approach for Optimal Model Reduction of Biomolecules
- PMID: 33108737
- PMCID: PMC7659038
- DOI: 10.1021/acs.jctc.0c00676
An Information-Theory-Based Approach for Optimal Model Reduction of Biomolecules
Abstract
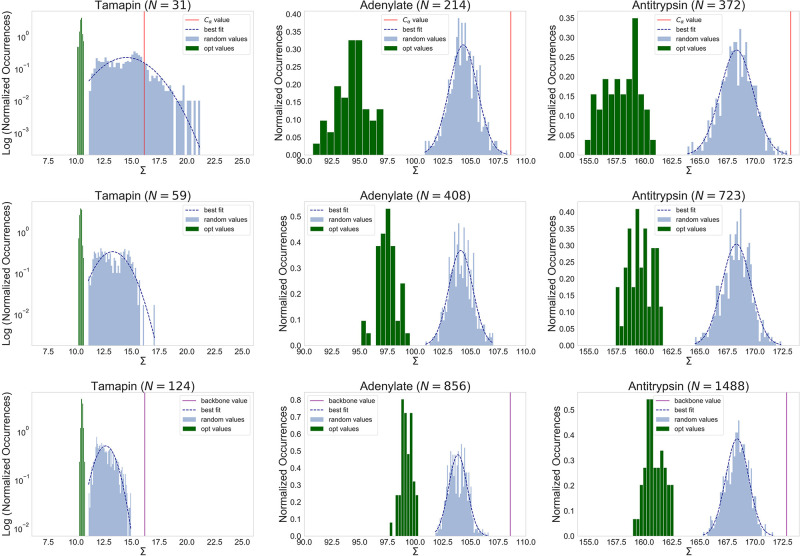
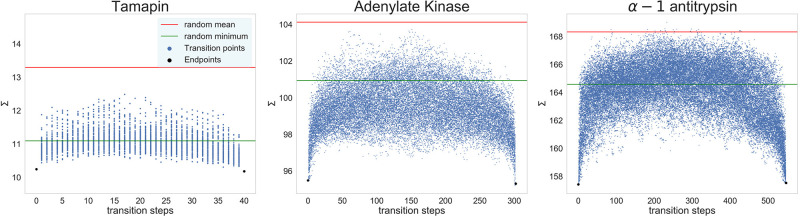
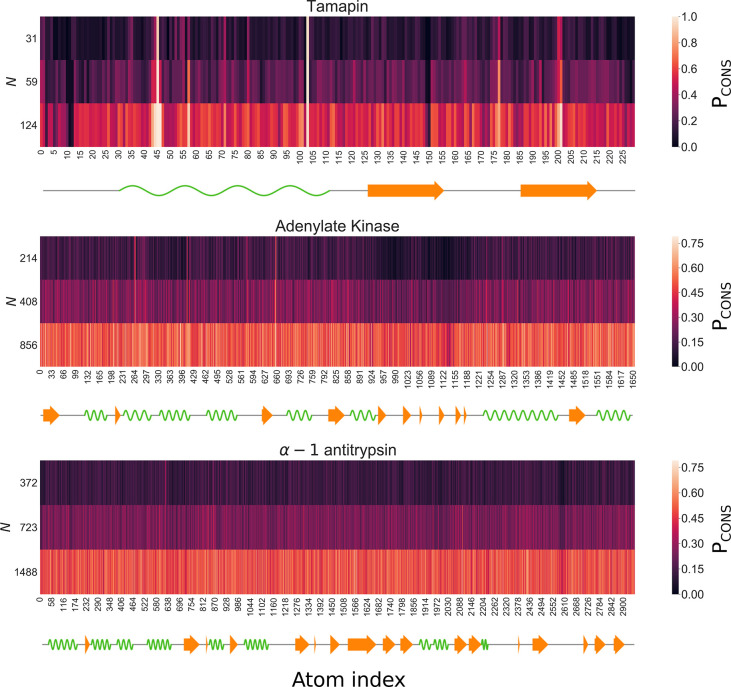
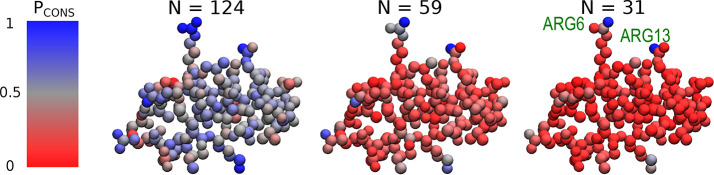
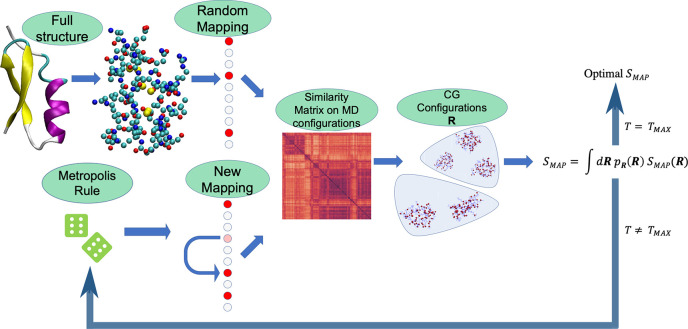
In theoretical modeling of a physical system, a crucial step consists of the identification of those degrees of freedom that enable a synthetic yet informative representation of it. While in some cases this selection can be carried out on the basis of intuition and experience, straightforward discrimination of the important features from the negligible ones is difficult for many complex systems, most notably heteropolymers and large biomolecules. We here present a thermodynamics-based theoretical framework to gauge the effectiveness of a given simplified representation by measuring its information content. We employ this method to identify those reduced descriptions of proteins, in terms of a subset of their atoms, that retain the largest amount of information from the original model; we show that these highly informative representations share common features that are intrinsically related to the biological properties of the proteins under examination, thereby establishing a bridge between protein structure, energetics, and function.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Alder B. J.; Wainwright T. E. Studies in Molecular Dynamics. I. General Method. J. Chem. Phys. 1959, 31, 459–466. 10.1063/1.1730376. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
