

Lost and Found: Re-searching and Re-scoring Proteomics Data Aids Genome Annotation and Improves Proteome Coverage
- PMID: 33109751
- PMCID: PMC7593589
- DOI: 10.1128/mSystems.00833-20
Lost and Found: Re-searching and Re-scoring Proteomics Data Aids Genome Annotation and Improves Proteome Coverage
Abstract
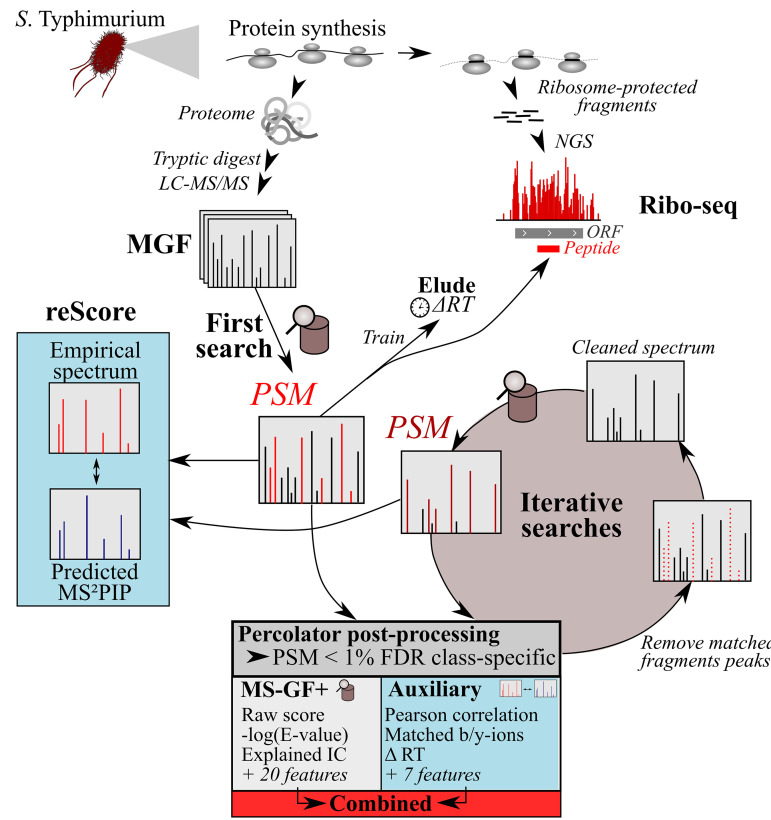
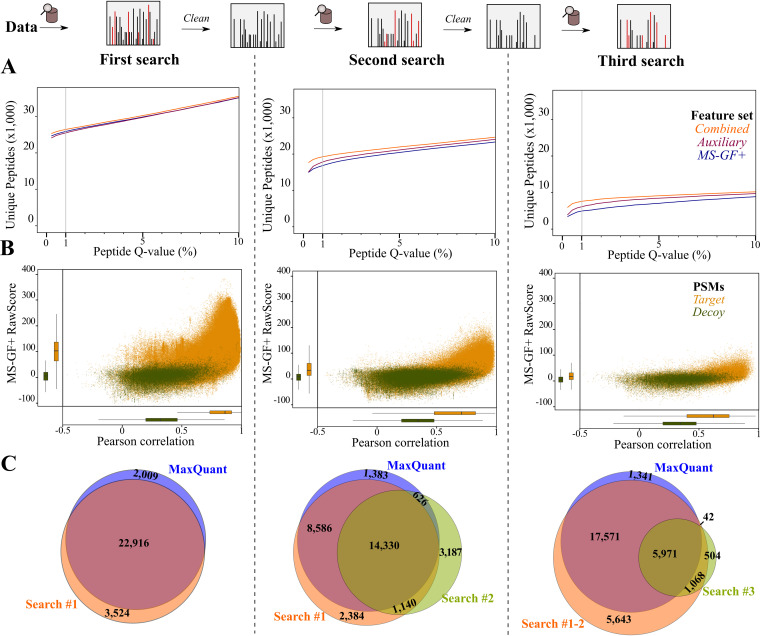
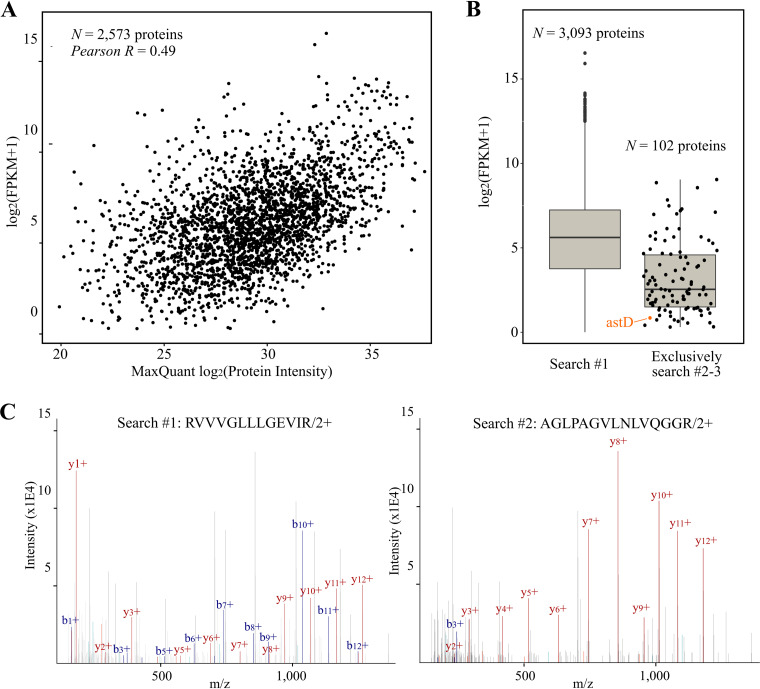
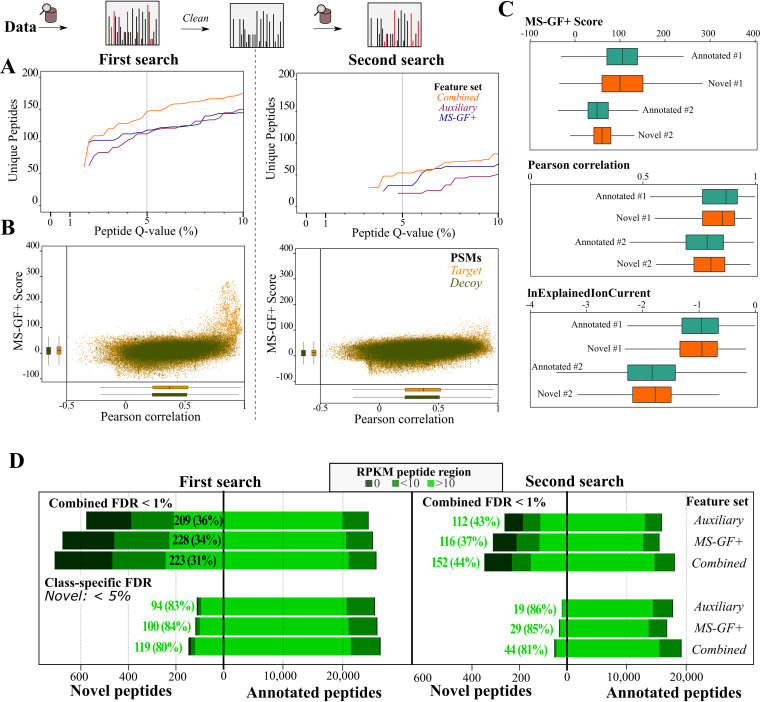
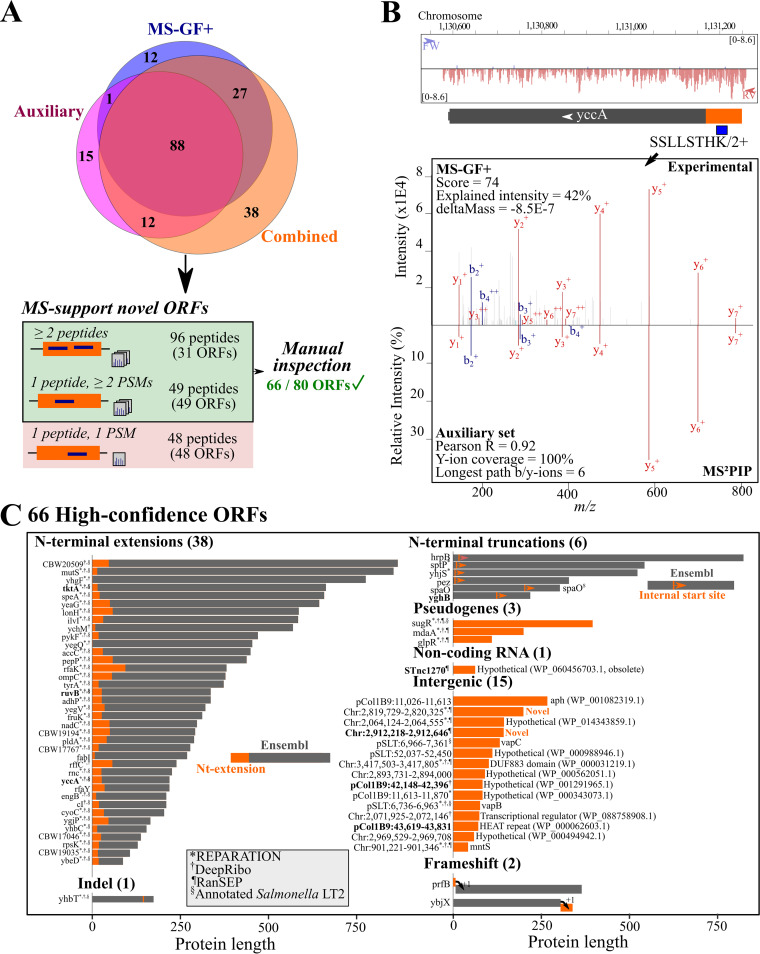
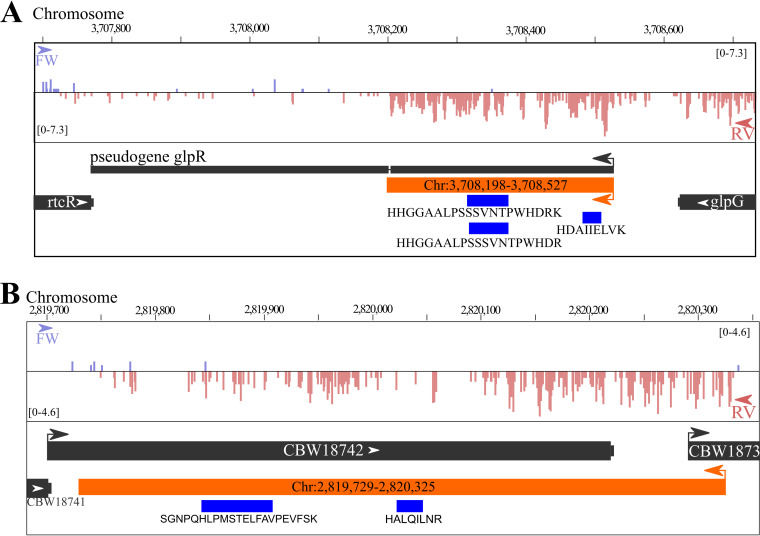
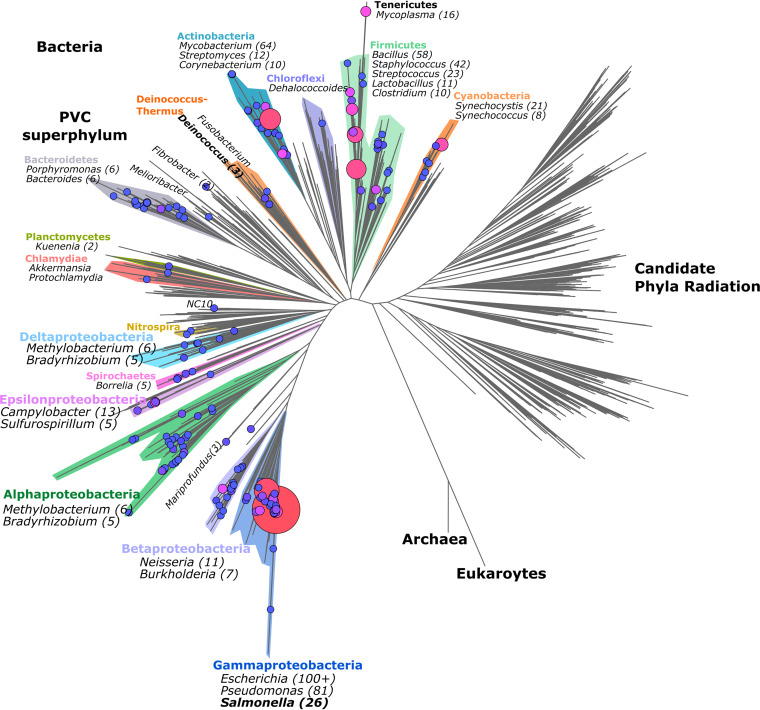
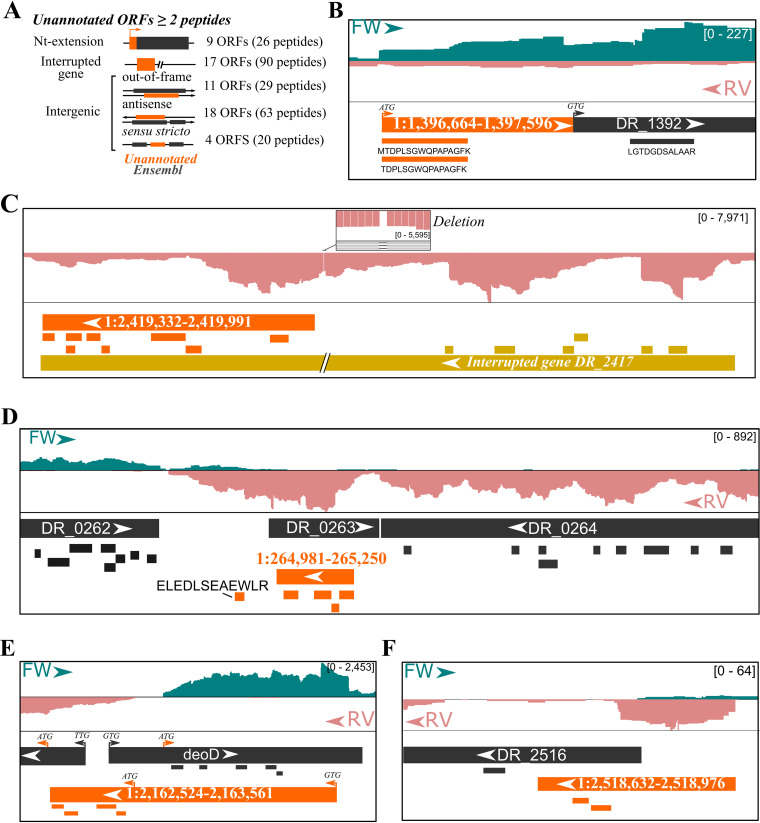
Prokaryotic genome annotation is heavily dependent on automated gene annotation pipelines that are prone to propagate errors and underestimate genome complexity. We describe an optimized proteogenomic workflow that uses ribosome profiling (ribo-seq) and proteomic data for Salmonella enterica serovar Typhimurium to identify unannotated proteins or alternative protein forms. This data analysis encompasses the searching of cofragmenting peptides and postprocessing with extended peptide-to-spectrum quality features, including comparison to predicted fragment ion intensities. When this strategy is applied, an enhanced proteome depth is achieved, as well as greater confidence for unannotated peptide hits. We demonstrate the general applicability of our pipeline by reanalyzing public Deinococcus radiodurans data sets. Taken together, our results show that systematic reanalysis using available prokaryotic (proteome) data sets holds great promise to assist in experimentally based genome annotation.IMPORTANCE Delineation of open reading frames (ORFs) causes persistent inconsistencies in prokaryote genome annotation. We demonstrate that by advanced (re)analysis of omics data, a higher proteome coverage and sensitive detection of unannotated ORFs can be achieved, which can be exploited for conditional bacterial genome (re)annotation, which is especially relevant in view of annotating the wealth of sequenced prokaryotic genomes obtained in recent years.
Keywords: Deinococcus radiodurans; Salmonella; alternative translation initiation; bacterial genome (re)annotation; chimeric spectra; riboproteogenomics; spectral re-scoring.
Copyright © 2020 Willems et al.
Figures
References
-
- Haft DH, DiCuccio M, Badretdin A, Brover V, Chetvernin V, O’Neill K, Li W, Chitsaz F, Derbyshire MK, Gonzales NR, Gwadz M, Lu F, Marchler GH, Song JS, Thanki N, Yamashita RA, Zheng C, Thibaud-Nissen F, Geer LY, Marchler-Bauer A, Pruitt KD. 2018. RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res 46:D851–D860. doi: 10.1093/nar/gkx1068. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
