

Spatiotemporal identification of druggable binding sites using deep learning
- PMID: 33110179
- PMCID: PMC7591901
- DOI: 10.1038/s42003-020-01350-0
Spatiotemporal identification of druggable binding sites using deep learning
Abstract
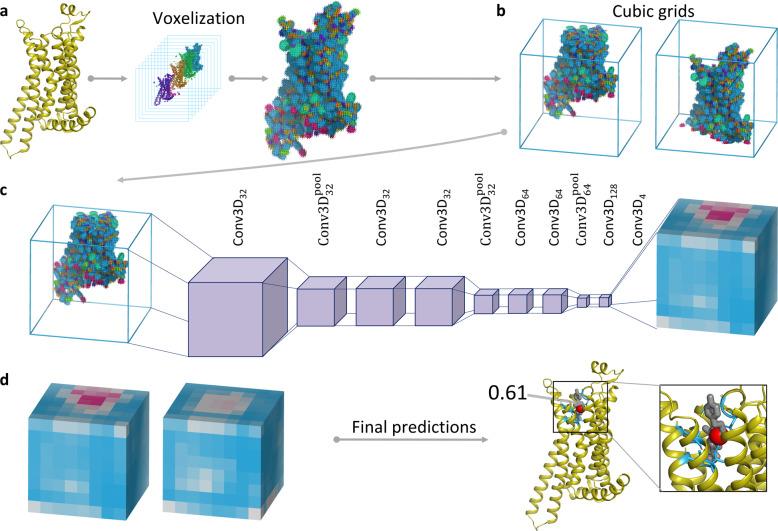
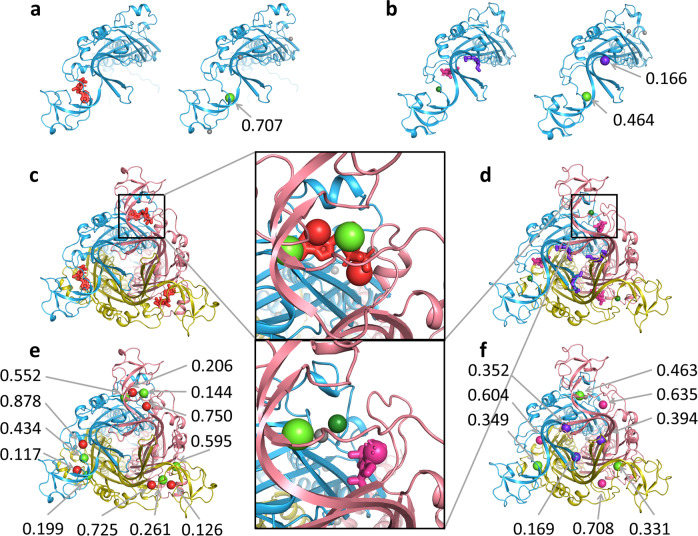
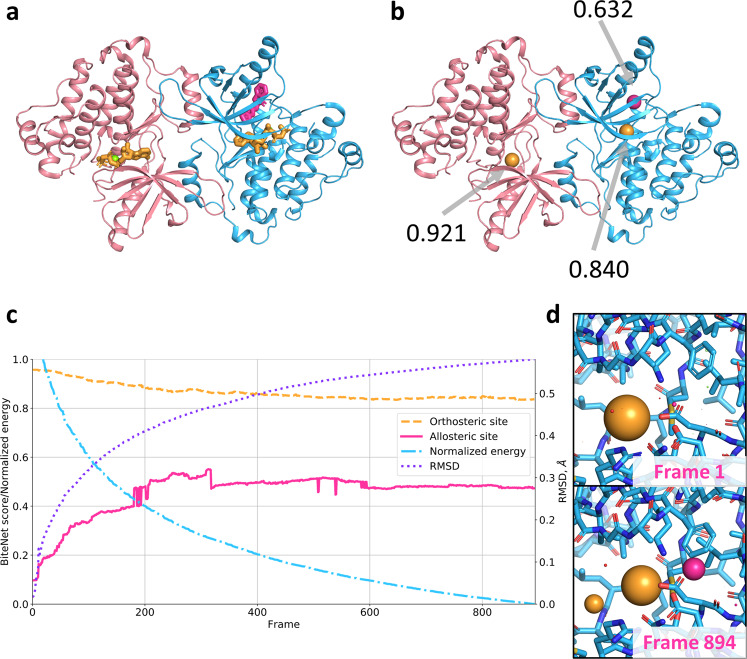
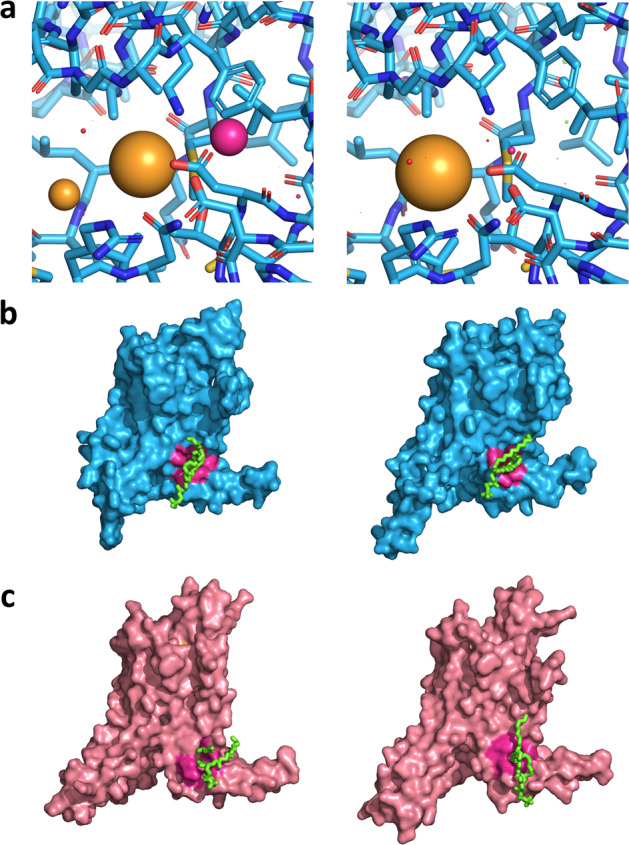
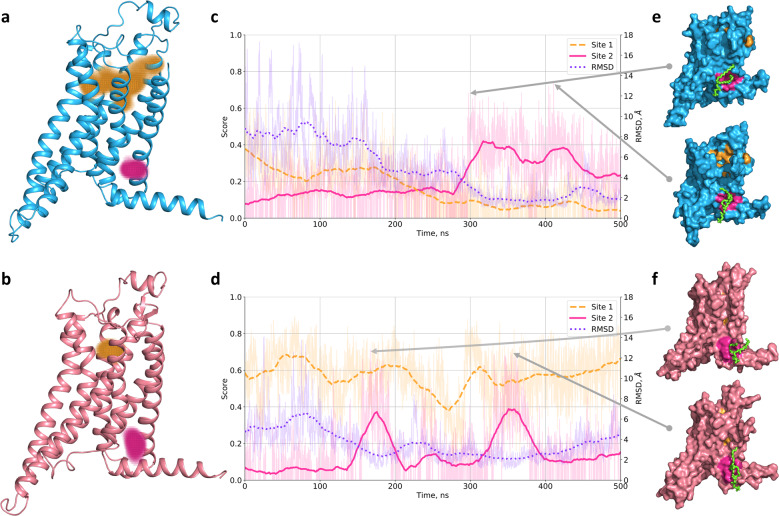
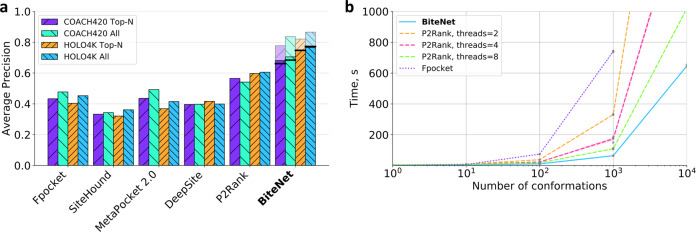
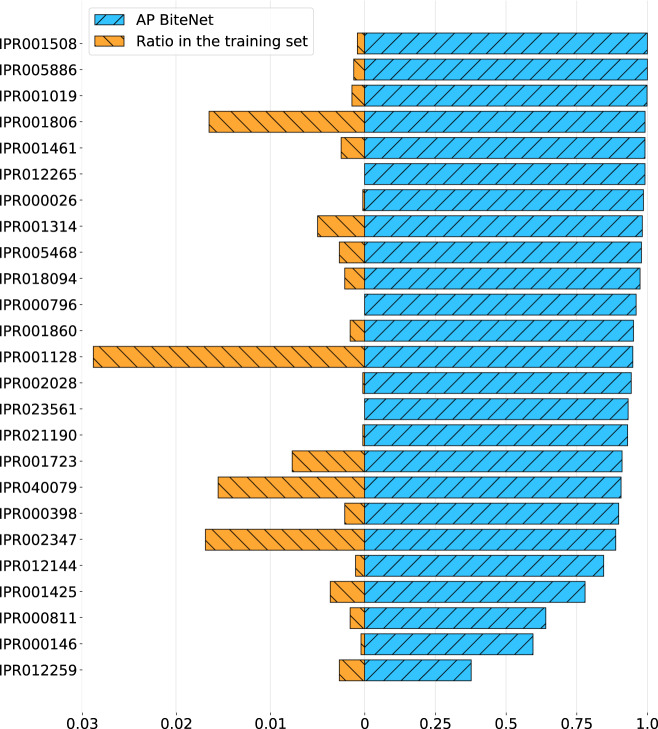
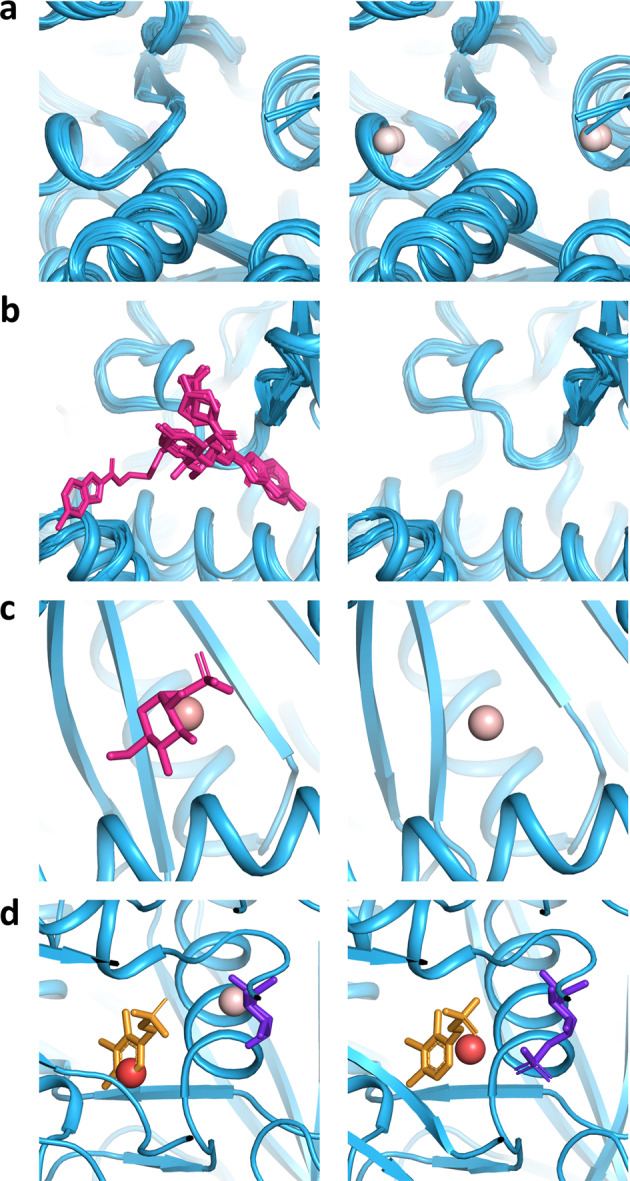
Identification of novel protein binding sites expands druggable genome and opens new opportunities for drug discovery. Generally, presence or absence of a binding site depends on the three-dimensional conformation of a protein, making binding site identification resemble the object detection problem in computer vision. Here we introduce a computational approach for the large-scale detection of protein binding sites, that considers protein conformations as 3D-images, binding sites as objects on these images to detect, and conformational ensembles of proteins as 3D-videos to analyze. BiteNet is suitable for spatiotemporal detection of hard-to-spot allosteric binding sites, as we showed for conformation-specific binding site of the epidermal growth factor receptor, oligomer-specific binding site of the ion channel, and binding site in G protein-coupled receptor. BiteNet outperforms state-of-the-art methods both in terms of accuracy and speed, taking about 1.5 minutes to analyze 1000 conformations of a protein with ~2000 atoms.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
