

CHARMM-GUI Free Energy Calculator for Absolute and Relative Ligand Solvation and Binding Free Energy Simulations
- PMID: 33112150
- PMCID: PMC7658063
- DOI: 10.1021/acs.jctc.0c00884
CHARMM-GUI Free Energy Calculator for Absolute and Relative Ligand Solvation and Binding Free Energy Simulations
Abstract
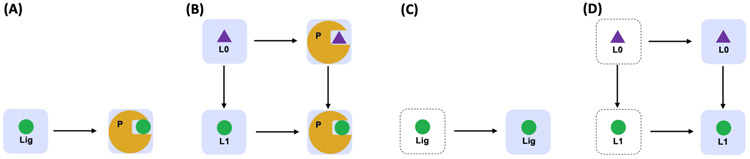
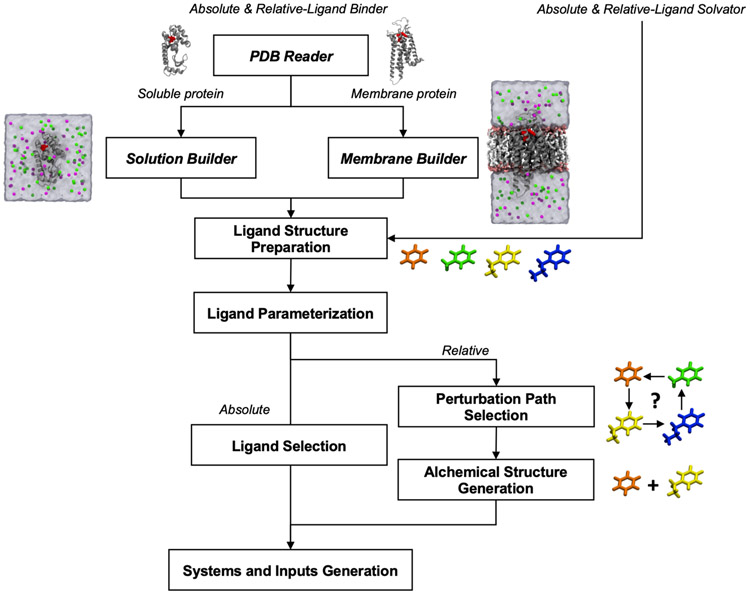
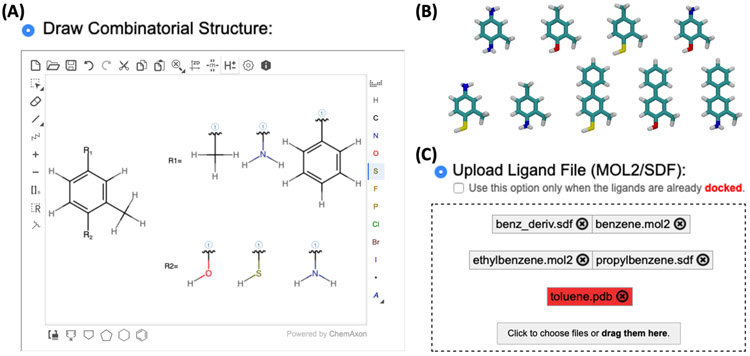
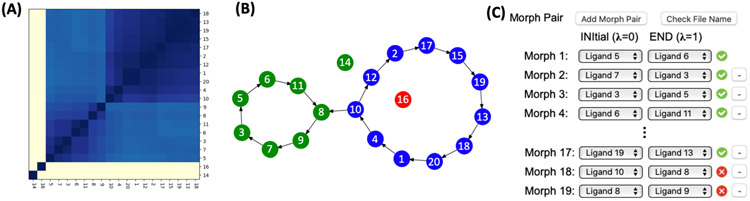
Alchemical free energy simulations have long been utilized to predict free energy changes for binding affinity and solubility of small molecules. However, while the theoretical foundation of these methods is well established, seamlessly handling many of the practical aspects regarding the preparation of the different thermodynamic end states of complex molecular systems and the numerous processing scripts often remains a burden for successful applications. In this work, we present CHARMM-GUI Free Energy Calculator (http://www.charmm-gui.org/input/fec) that provides various alchemical free energy perturbation molecular dynamics (FEP/MD) systems with input and post-processing scripts for NAMD and GENESIS. Four submodules are available: Absolute Ligand Binder (for absolute ligand binding FEP/MD), Relative Ligand Binder (for relative ligand binding FEP/MD), Absolute Ligand Solvator (for absolute ligand solvation FEP/MD), and Relative Ligand Solvator (for relative ligand solvation FEP/MD). Each module is designed to build multiple systems of a set of selected ligands at once for high-throughput FEP/MD simulations. The capability of Free Energy Calculator is illustrated by absolute and relative solvation FEP/MD of a set of ligands and absolute and relative binding FEP/MD of a set of ligands for T4-lysozyme in solution and the adenosine A2A receptor in a membrane. The calculated free energy values are overall consistent with the experimental and published free energy results (within ∼1 kcal/mol). We hope that Free Energy Calculator is useful to carry out high-throughput FEP/MD simulations in the field of biomolecular sciences and drug discovery.
Figures
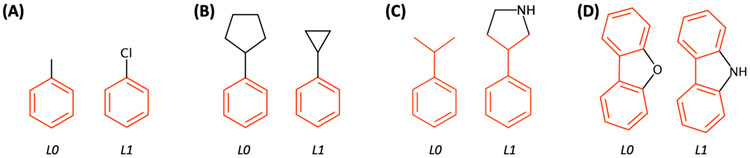
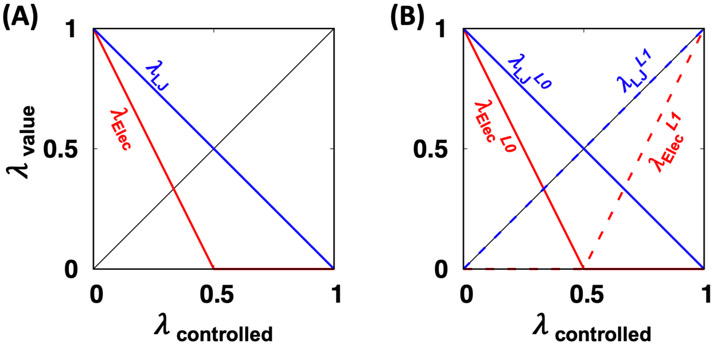
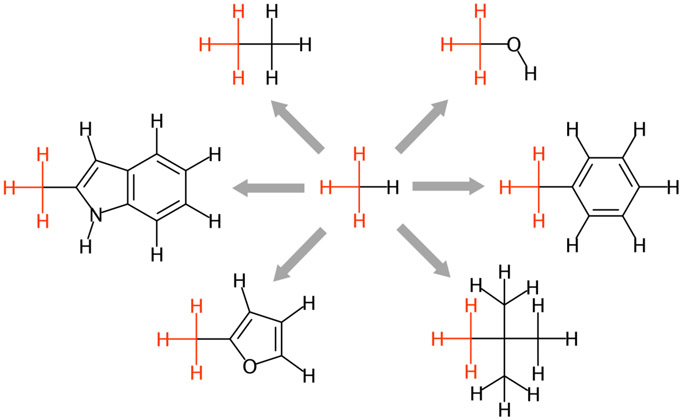
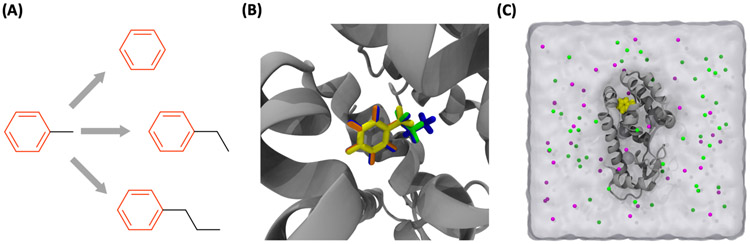
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources