

Widespread Transcriptional Readthrough Caused by Nab2 Depletion Leads to Chimeric Transcripts with Retained Introns
- PMID: 33113357
- PMCID: PMC7774305
- DOI: 10.1016/j.celrep.2020.108324
Widespread Transcriptional Readthrough Caused by Nab2 Depletion Leads to Chimeric Transcripts with Retained Introns
Erratum in
-
Widespread Transcriptional Readthrough Caused by Nab2 Depletion Leads to Chimeric Transcripts with Retained Introns.Cell Rep. 2020 Dec 29;33(13):108496. doi: 10.1016/j.celrep.2020.108496. Cell Rep. 2020. PMID: 33378663 Free PMC article. No abstract available.
Abstract
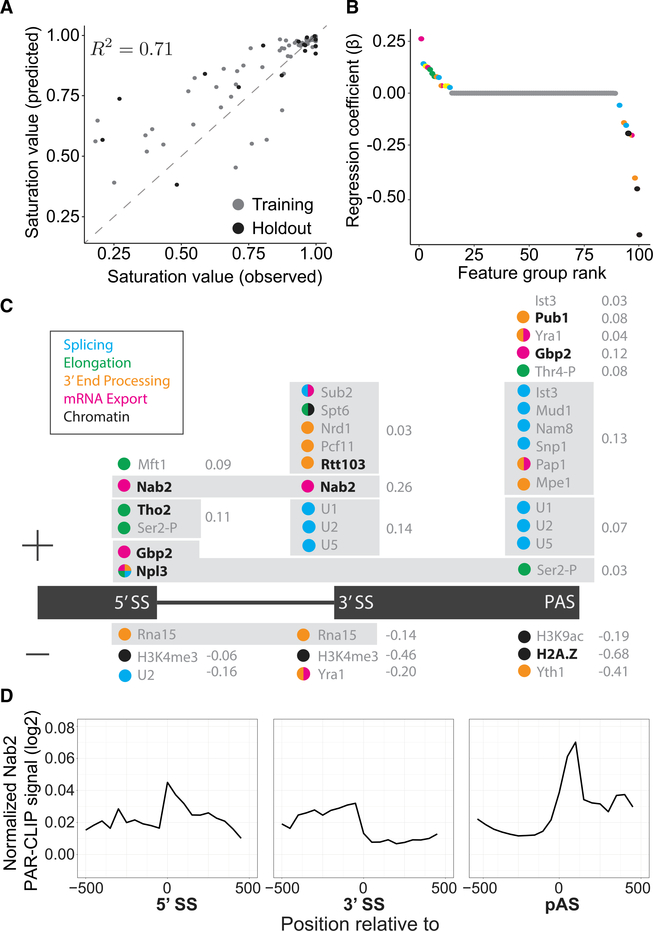
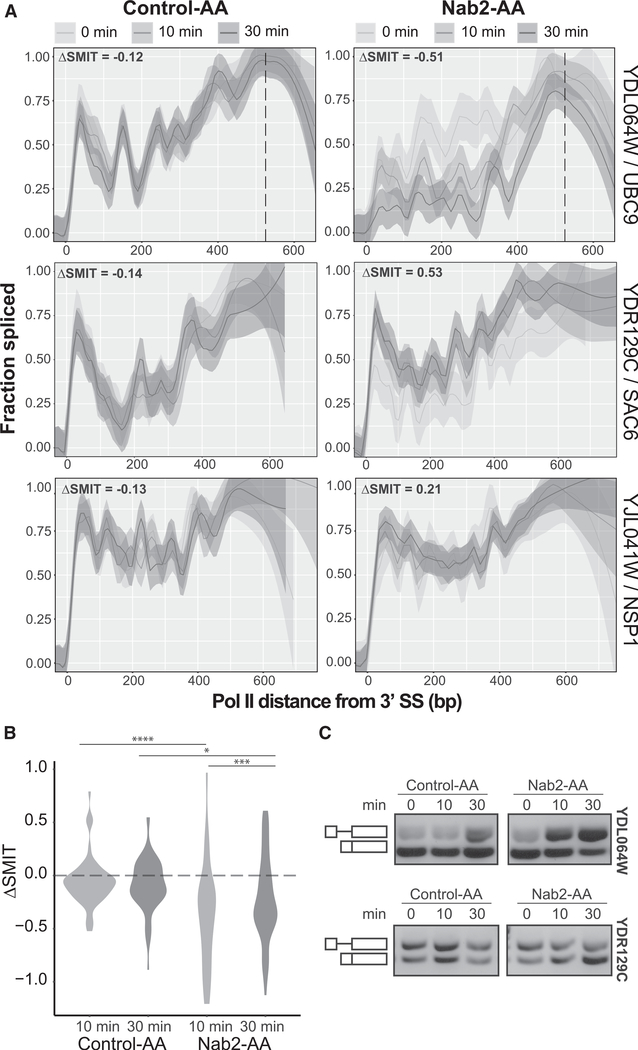
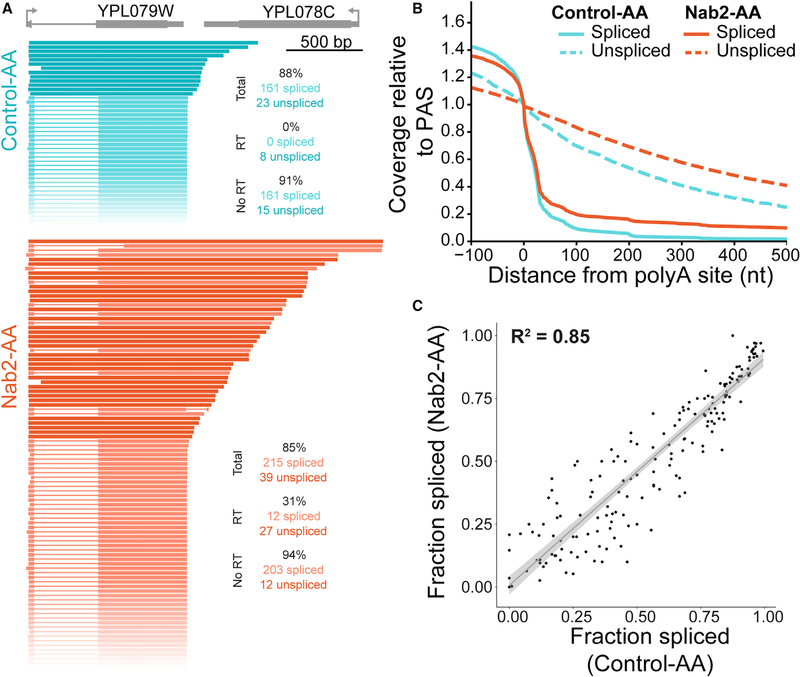
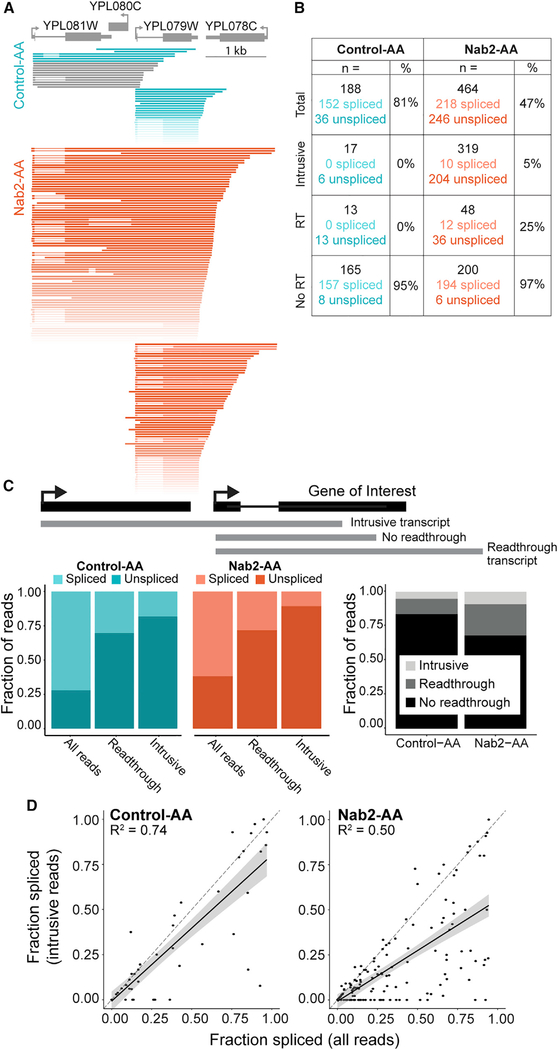
Nascent RNA sequencing has revealed that pre-mRNA splicing can occur shortly after introns emerge from RNA polymerase II (RNA Pol II). Differences in co-transcriptional splicing profiles suggest regulation by cis- and/or trans-acting factors. Here, we use single-molecule intron tracking (SMIT) to identify a cohort of regulators by machine learning in budding yeast. Of these, Nab2 displays reduced co-transcriptional splicing when depleted. Unexpectedly, these splicing defects are attributable to aberrant "intrusive" transcriptional readthrough from upstream genes, as revealed by long-read sequencing. Transcripts that originate from the intron-containing gene's own transcription start site (TSS) are efficiently spliced, indicating no direct role of Nab2 in splicing per se. This work highlights the coupling between transcription, splicing, and 3' end formation in the context of gene organization along chromosomes. We conclude that Nab2 is required for proper 3' end processing, which ensures gene-specific control of co-transcriptional RNA processing.
Keywords: 3′ end cleavage; SMIT; co-transcriptional processes; intrusive transcripts; long-read sequencing; machine learning; nascent RNA; pre-mRNA splicing; transcriptional readthrough.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no conflict of interest.
Figures
References
-
- Aibara S, Gordon JMB, Riesterer AS, McLaughlin SH, and Stewart M (2017). Structural basis for the dimerization of Nab2 generated by RNA binding provides insight into its contribution to both poly(A) tail length determination and transcript compaction in Saccharomyces cerevisiae. Nucleic Acids Res. 45, 1529–1538. - PMC - PubMed
-
- Baejen C, Torkler P, Gressel S, Essig K, Söding J, and Cramer P. (2014). Transcriptome maps of mRNP biogenesis factors define pre-mRNA recognition. Mol. Cell 55, 745–757. - PubMed
-
- Baejen C, Andreani J, Torkler P, Battaglia S, Schwalb B, Lidschreiber M, Maier KC, Boltendahl A, Rus P, Esslinger S, et al. (2017). Genome-wide Analysis of RNA Polymerase II Termination at Protein-Coding Genes. Mol. Cell 66, 38–49.e6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
