

Detection and Characterization of Swine Origin Influenza A(H1N1) Pandemic 2009 Viruses in Humans following Zoonotic Transmission
- PMID: 33115872
- PMCID: PMC7944445
- DOI: 10.1128/JVI.01066-20
Detection and Characterization of Swine Origin Influenza A(H1N1) Pandemic 2009 Viruses in Humans following Zoonotic Transmission
Abstract
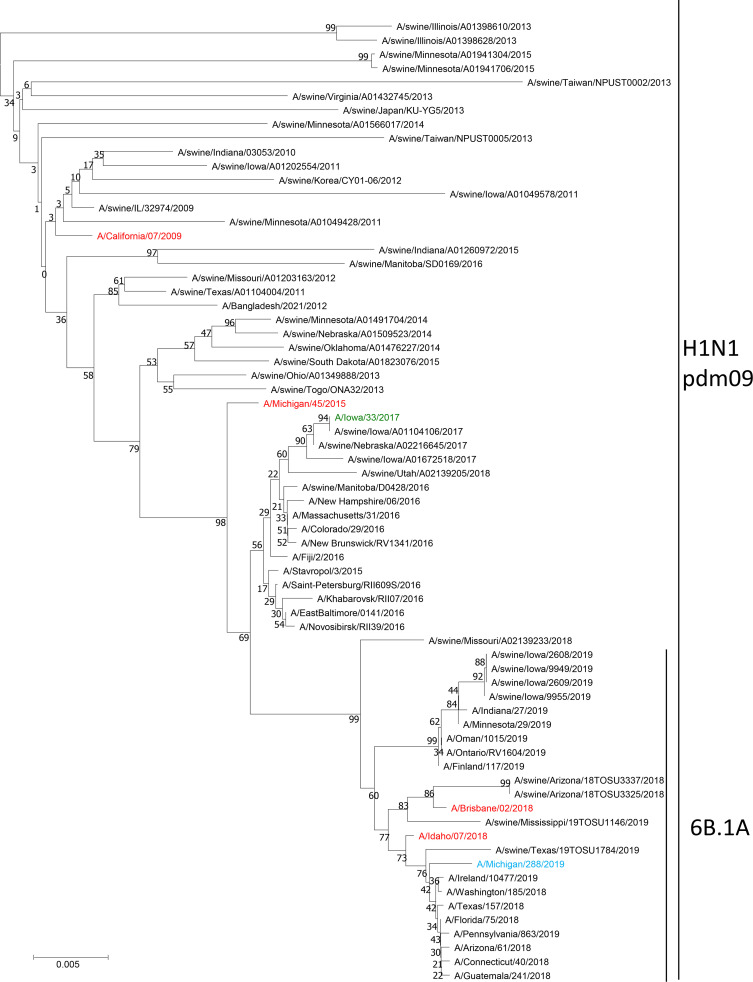
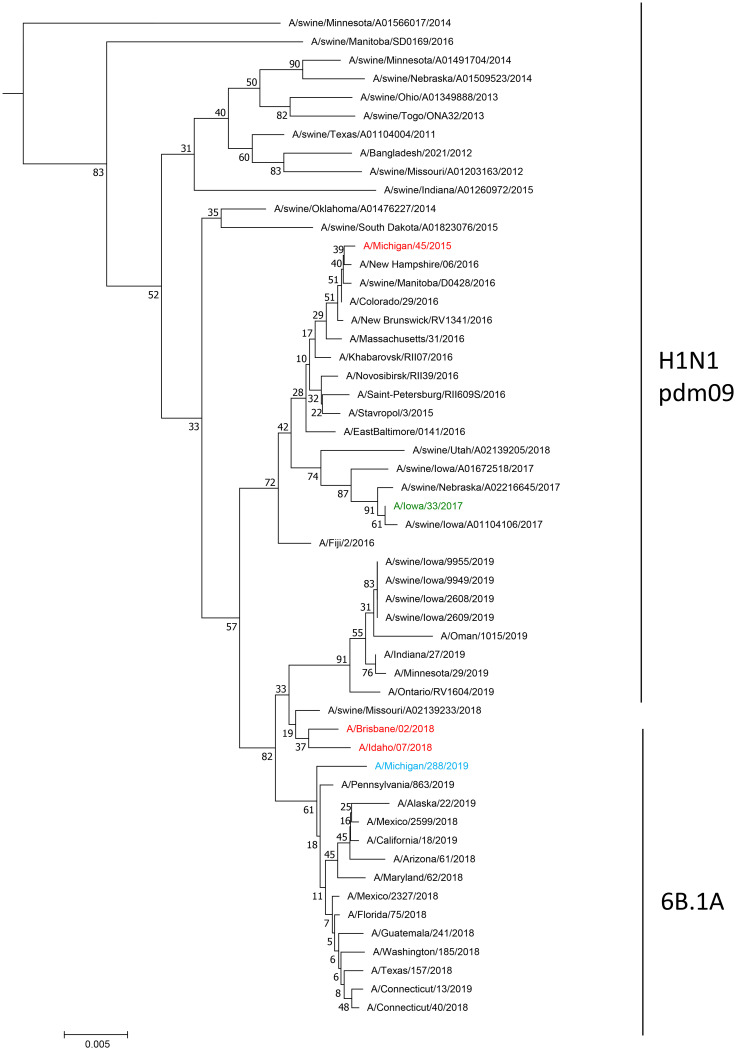
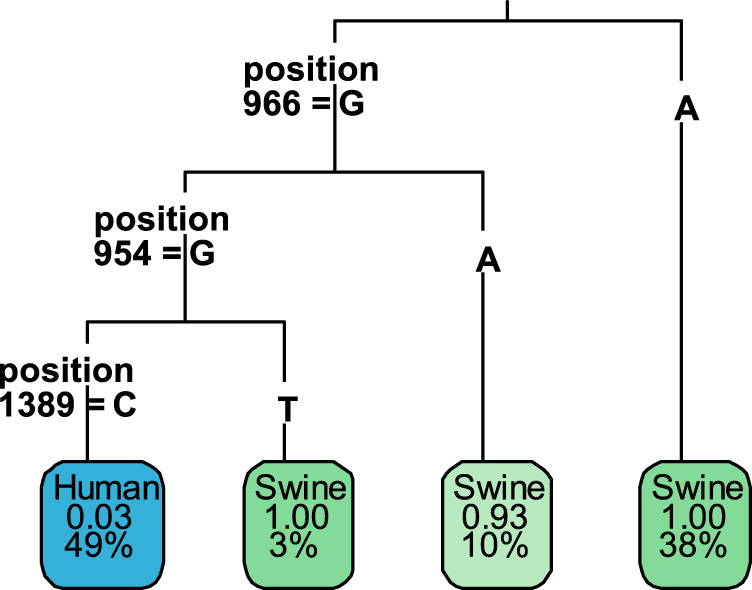
Human-to-swine transmission of seasonal influenza viruses has led to sustained human-like influenza viruses circulating in the U.S. swine population. While some reverse zoonotic-origin viruses adapt and become enzootic in swine, nascent reverse zoonoses may result in virus detections that are difficult to classify as "swine-origin" or "human-origin" due to the genetic similarity of circulating viruses. This is the case for human-origin influenza A(H1N1) pandemic 2009 (pdm09) viruses detected in pigs following numerous reverse zoonosis events since the 2009 pandemic. We report the identification of two human infections with A(H1N1)pdm09 viruses originating from swine hosts and classify them as "swine-origin" variant influenza viruses based on phylogenetic analysis and sequence comparison methods. Phylogenetic analyses of viral genomes from two cases revealed these viruses were reassortants containing A(H1N1)pdm09 hemagglutinin (HA) and neuraminidase (NA) genes with genetic combinations derived from the triple reassortant internal gene cassette. Follow-up investigations determined that one individual had direct exposure to swine in the week preceding illness onset, while another did not report swine exposure. The swine-origin A(H1N1) variant cases were resolved by full genome sequence comparison of the variant viruses to swine influenza genomes. However, if reassortment does not result in the acquisition of swine-associated genes and swine virus genomic sequences are not available from the exposure source, future cases may not be discernible. We have developed a pipeline that performs maximum likelihood analyses, a k-mer-based set difference algorithm, and random forest algorithms to identify swine-associated sequences in the hemagglutinin gene to differentiate between human-origin and swine-origin A(H1N1)pdm09 viruses.IMPORTANCE Influenza virus infects a wide range of hosts, resulting in illnesses that vary from asymptomatic cases to severe pneumonia and death. Viral transfer can occur between human and nonhuman hosts, resulting in human and nonhuman origin viruses circulating in novel hosts. In this work, we have identified the first case of a swine-origin influenza A(H1N1)pdm09 virus resulting in a human infection. This shows that these viruses not only circulate in swine hosts, but are continuing to evolve and distinguish themselves from previously circulating human-origin influenza viruses. The development of techniques for distinguishing human-origin and swine-origin viruses are necessary for the continued surveillance of influenza viruses. We show that unique genetic signatures can differentiate circulating swine-associated strains from circulating human-associated strains of influenza A(H1N1)pdm09, and these signatures can be used to enhance surveillance of swine-origin influenza.
Keywords: A(H1N1)pdm09; influenza; pandemic; random forest; swine-origin; zoonotic transmission.
Copyright © 2020 American Society for Microbiology.
Figures

References
-
- Simon G, Larsen LE, Dürrwald R, Foni E, Harder T, Van Reeth K, Markowska-Daniel I, Reid SM, Dan A, Maldonado J, Huovilainen A, Billinis C, Davidson I, Agüero M, Vila T, Hervé S, Breum SØ, Chiapponi C, Urbaniak K, Kyriakis CS, Brown IH, Loeffen W, ESNIP3 consortium. 2014. European surveillance network for influenza in pigs: surveillance programs, diagnostic tools and swine influenza virus subtypes identified in 14 European countries from 2010 to 2013. PLoS One 9:e115815-21. doi: 10.1371/journal.pone.0115815. - DOI - PMC - PubMed
-
- Chastagner A, Hervé S, Bonin E, Quéguiner S, Hirchaud E, Henritzi D, Béven V, Gorin S, Barbier N, Blanchard Y, Simon G. 2018. Spatiotemporal distribution and evolution of the A/H1N1 2009 pandemic influenza virus in pigs in France from 2009 to 2017: identification of a potential swine-specific lineage. J Virol 92:440–442. doi: 10.1128/JVI.00988-18. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
