

High-depth African genomes inform human migration and health
- PMID: 33116287
- PMCID: PMC7759466
- DOI: 10.1038/s41586-020-2859-7
High-depth African genomes inform human migration and health
Erratum in
-
Author Correction: High-depth African genomes inform human migration and health.Nature. 2021 Apr;592(7856):E26. doi: 10.1038/s41586-021-03286-9. Nature. 2021. PMID: 33846614 Free PMC article. No abstract available.
Abstract
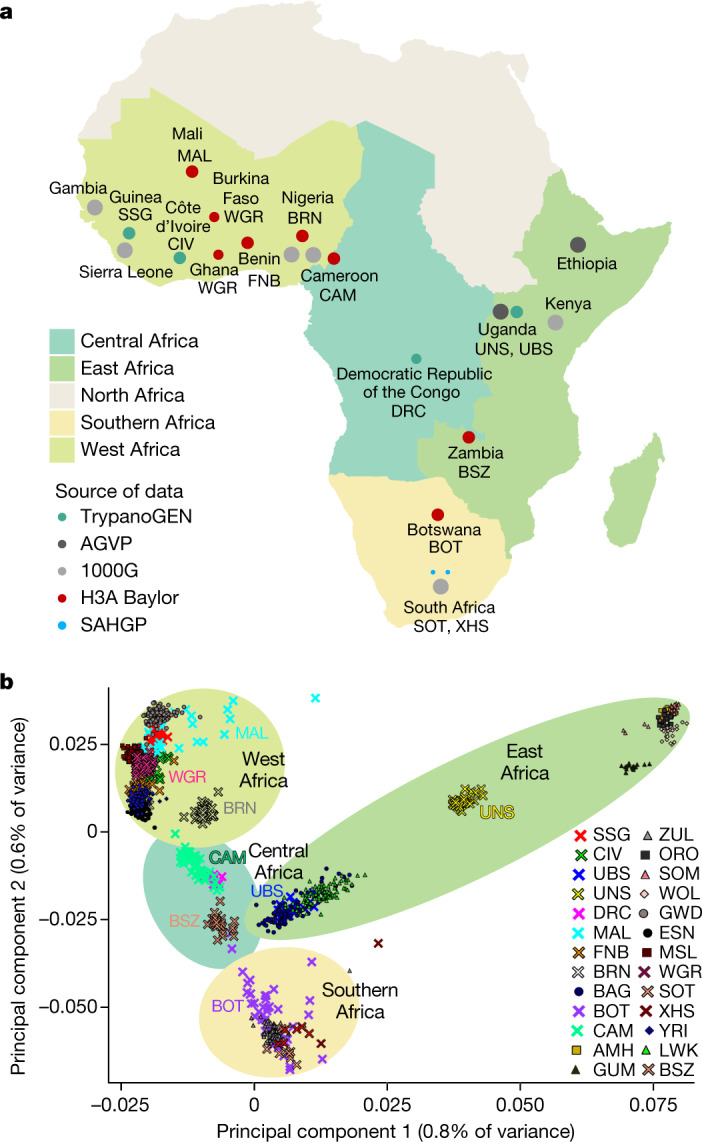
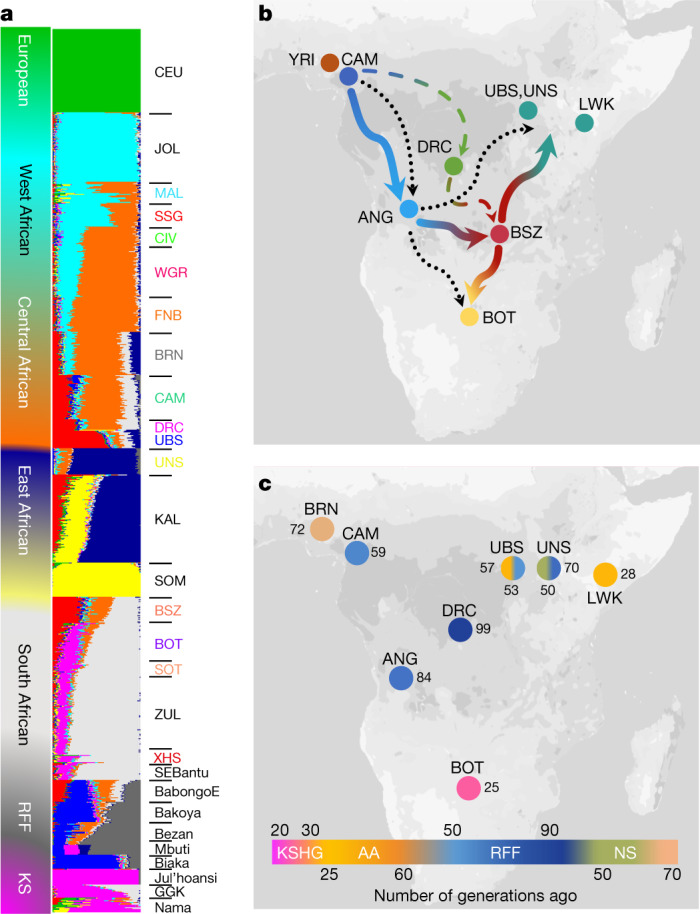
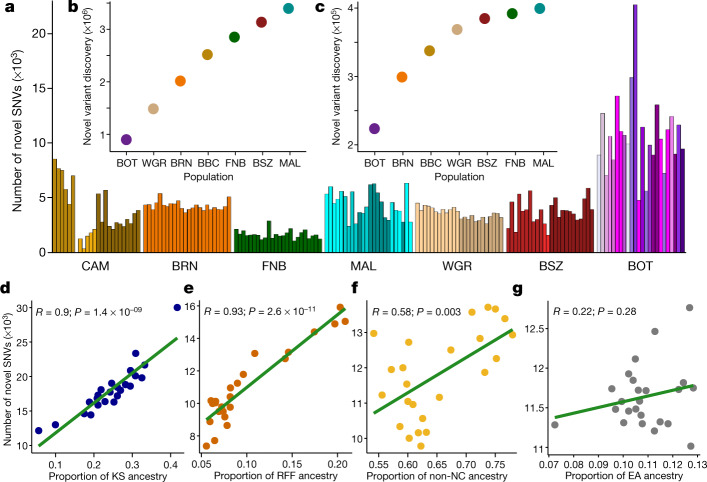
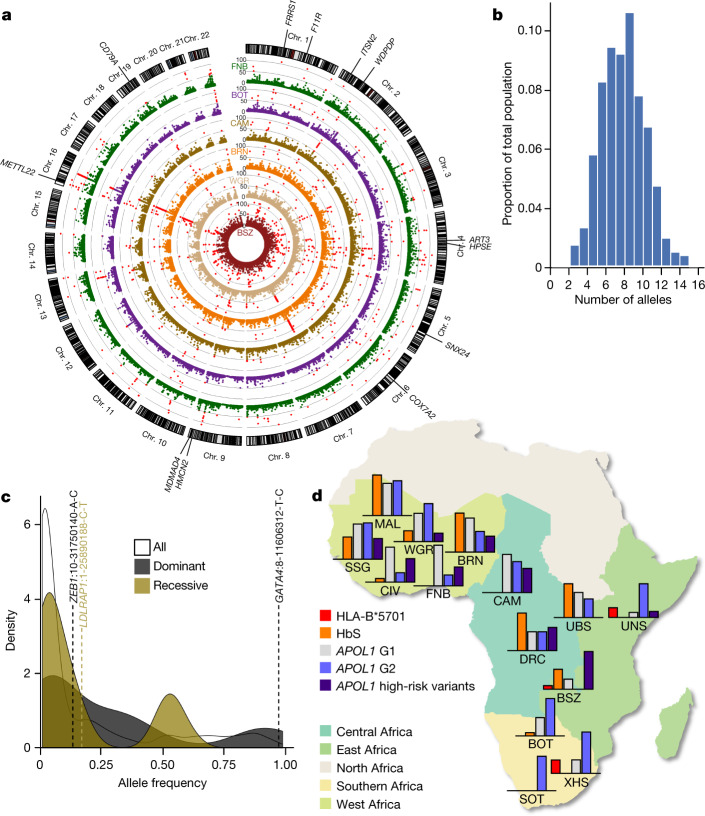
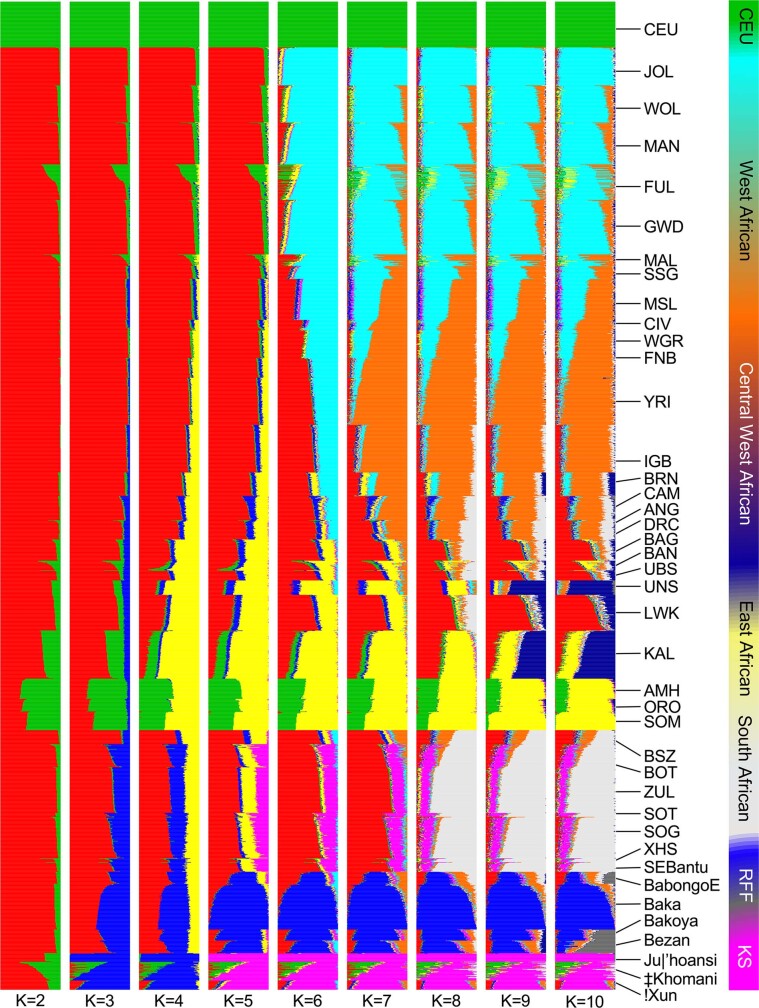
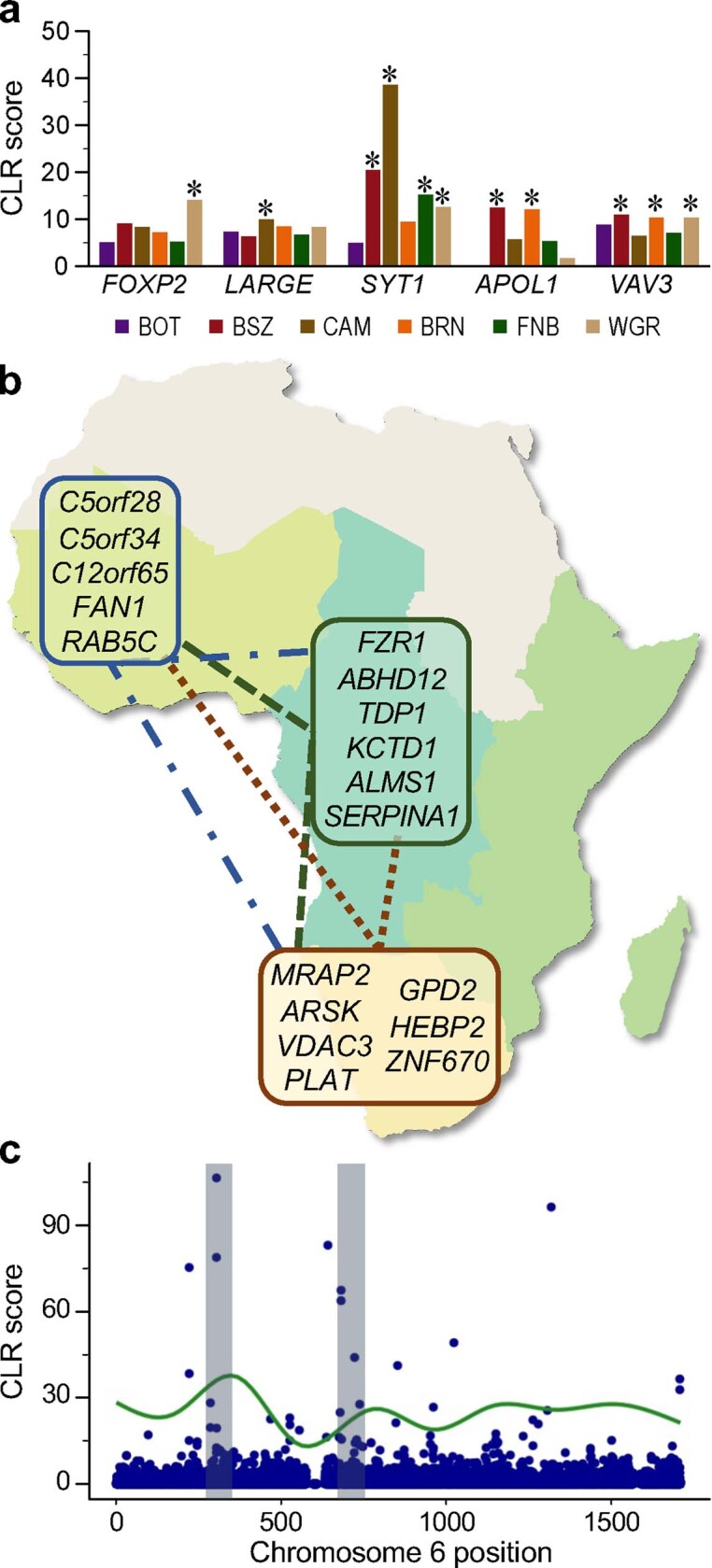
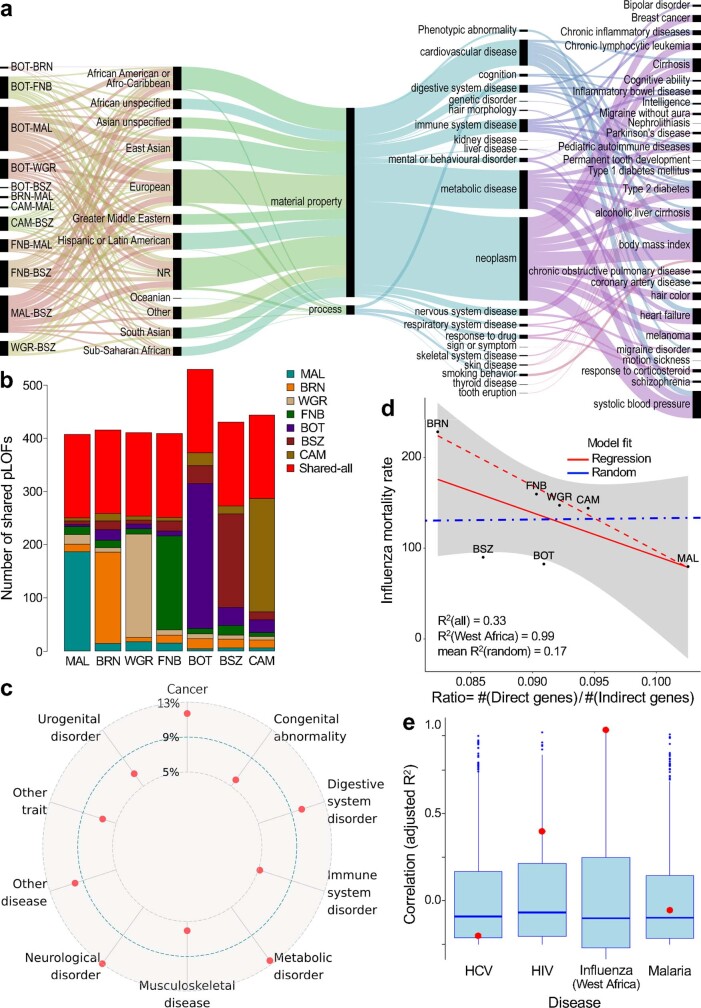
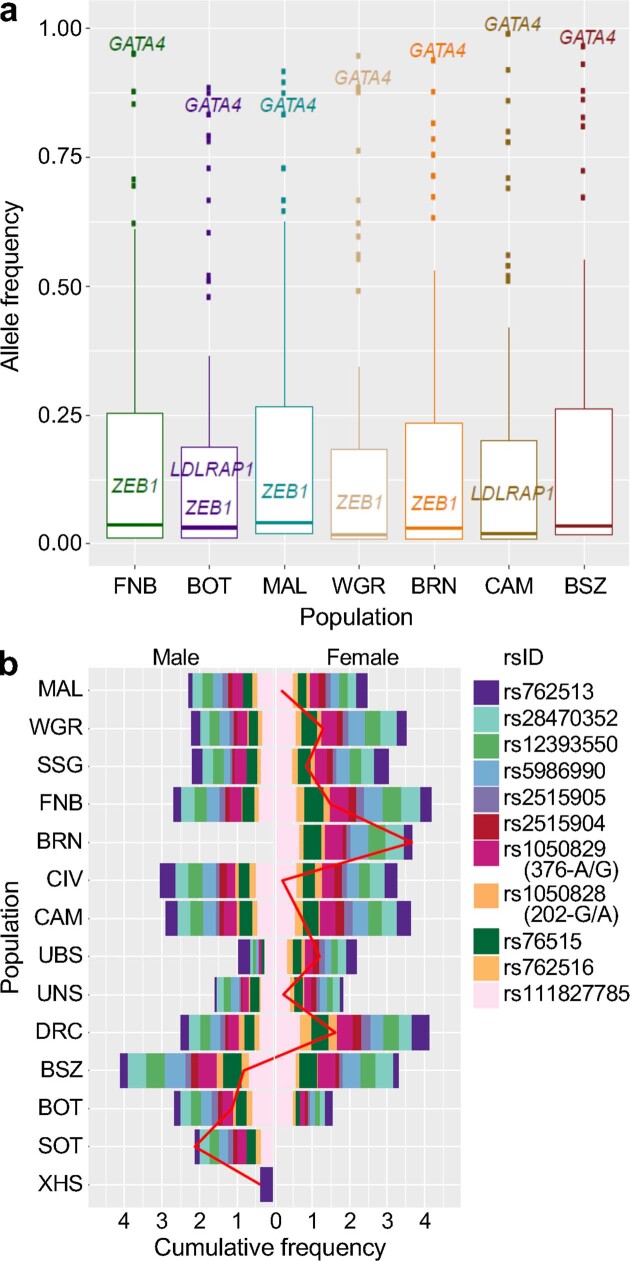
The African continent is regarded as the cradle of modern humans and African genomes contain more genetic variation than those from any other continent, yet only a fraction of the genetic diversity among African individuals has been surveyed1. Here we performed whole-genome sequencing analyses of 426 individuals-comprising 50 ethnolinguistic groups, including previously unsampled populations-to explore the breadth of genomic diversity across Africa. We uncovered more than 3 million previously undescribed variants, most of which were found among individuals from newly sampled ethnolinguistic groups, as well as 62 previously unreported loci that are under strong selection, which were predominantly found in genes that are involved in viral immunity, DNA repair and metabolism. We observed complex patterns of ancestral admixture and putative-damaging and novel variation, both within and between populations, alongside evidence that Zambia was a likely intermediate site along the routes of expansion of Bantu-speaking populations. Pathogenic variants in genes that are currently characterized as medically relevant were uncommon-but in other genes, variants denoted as 'likely pathogenic' in the ClinVar database were commonly observed. Collectively, these findings refine our current understanding of continental migration, identify gene flow and the response to human disease as strong drivers of genome-level population variation, and underscore the scientific imperative for a broader characterization of the genomic diversity of African individuals to understand human ancestry and improve health.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Africa's people must be able to write their own genomics agenda.Nature. 2020 Oct;586(7831):644. doi: 10.1038/d41586-020-03028-3. Nature. 2020. PMID: 33116292 No abstract available.
-
Embracing African Genetic Diversity.Med. 2021 Jan 15;2(1):19-20. doi: 10.1016/j.medj.2020.12.019. Med. 2021. PMID: 35590130
References
Publication types
MeSH terms
Grants and funding
- U24 HG006941/HG/NHGRI NIH HHS/United States
- U54 HG006947/HG/NHGRI NIH HHS/United States
- U01 HG007044/HG/NHGRI NIH HHS/United States
- U54 AI110398/AI/NIAID NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U54 DK116913/DK/NIDDK NIH HHS/United States
- U54 HG006938/HG/NHGRI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- U54 HG006939/HG/NHGRI NIH HHS/United States
- U01 HG007459/HG/NHGRI NIH HHS/United States
- U24 HL135600/HL/NHLBI NIH HHS/United States
- U01 MH115483/MH/NIMH NIH HHS/United States
- 099310/Z/12/Z/WT_/Wellcome Trust/United Kingdom
- U01 HG009716/HG/NHGRI NIH HHS/United States
