

Single-cell mutation analysis of clonal evolution in myeloid malignancies
- PMID: 33116311
- PMCID: PMC7677169
- DOI: 10.1038/s41586-020-2864-x
Single-cell mutation analysis of clonal evolution in myeloid malignancies
Abstract
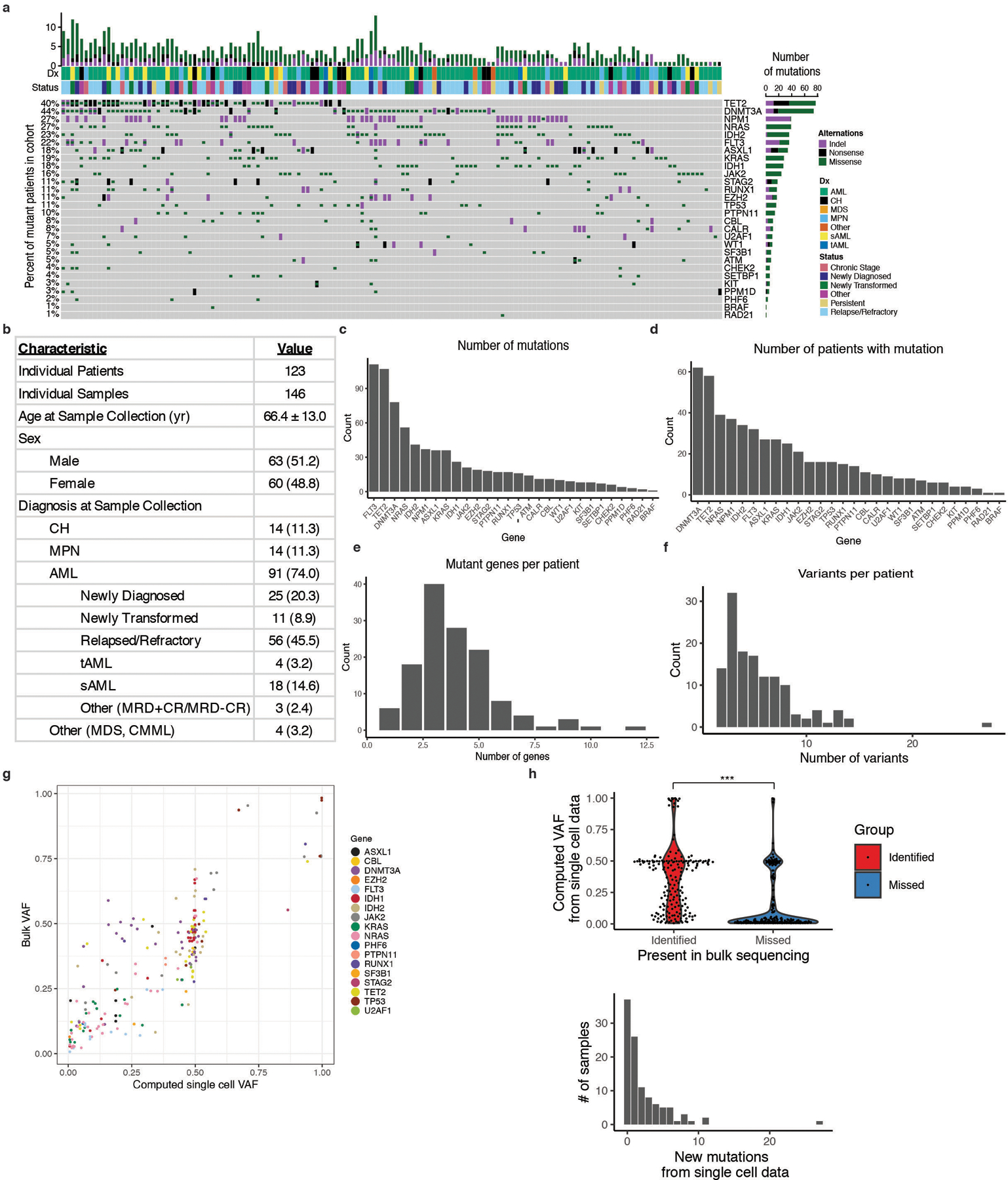
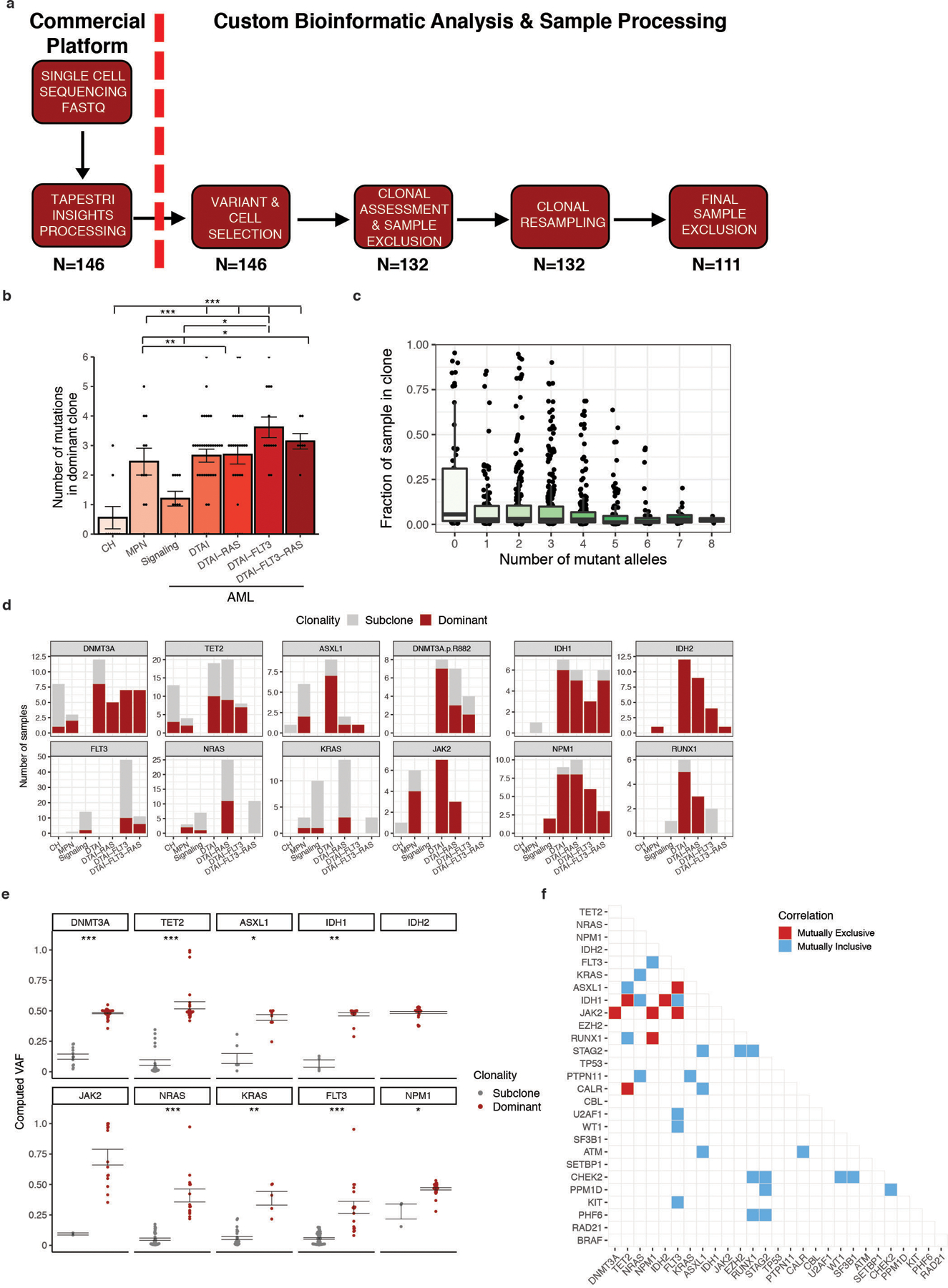
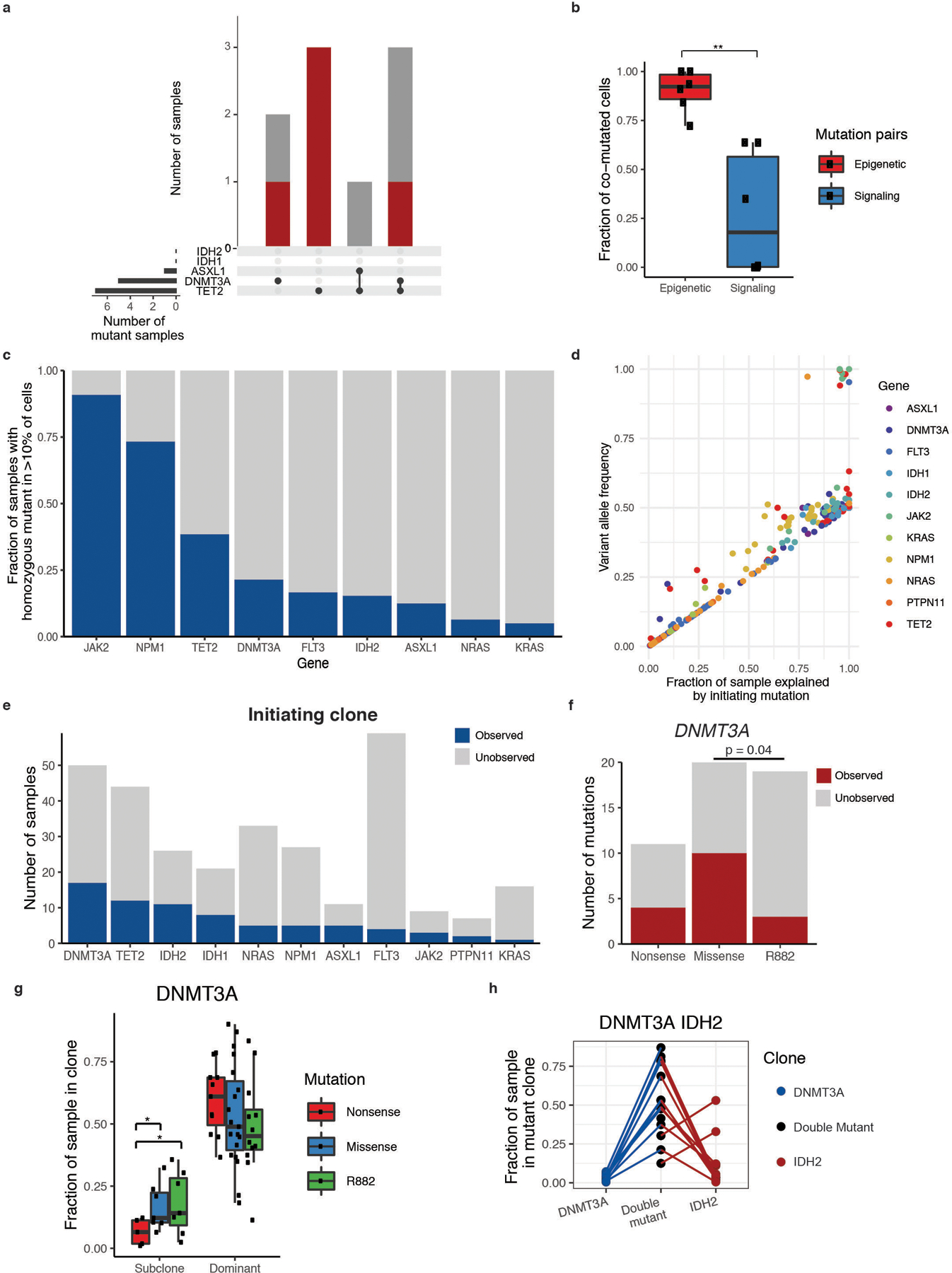
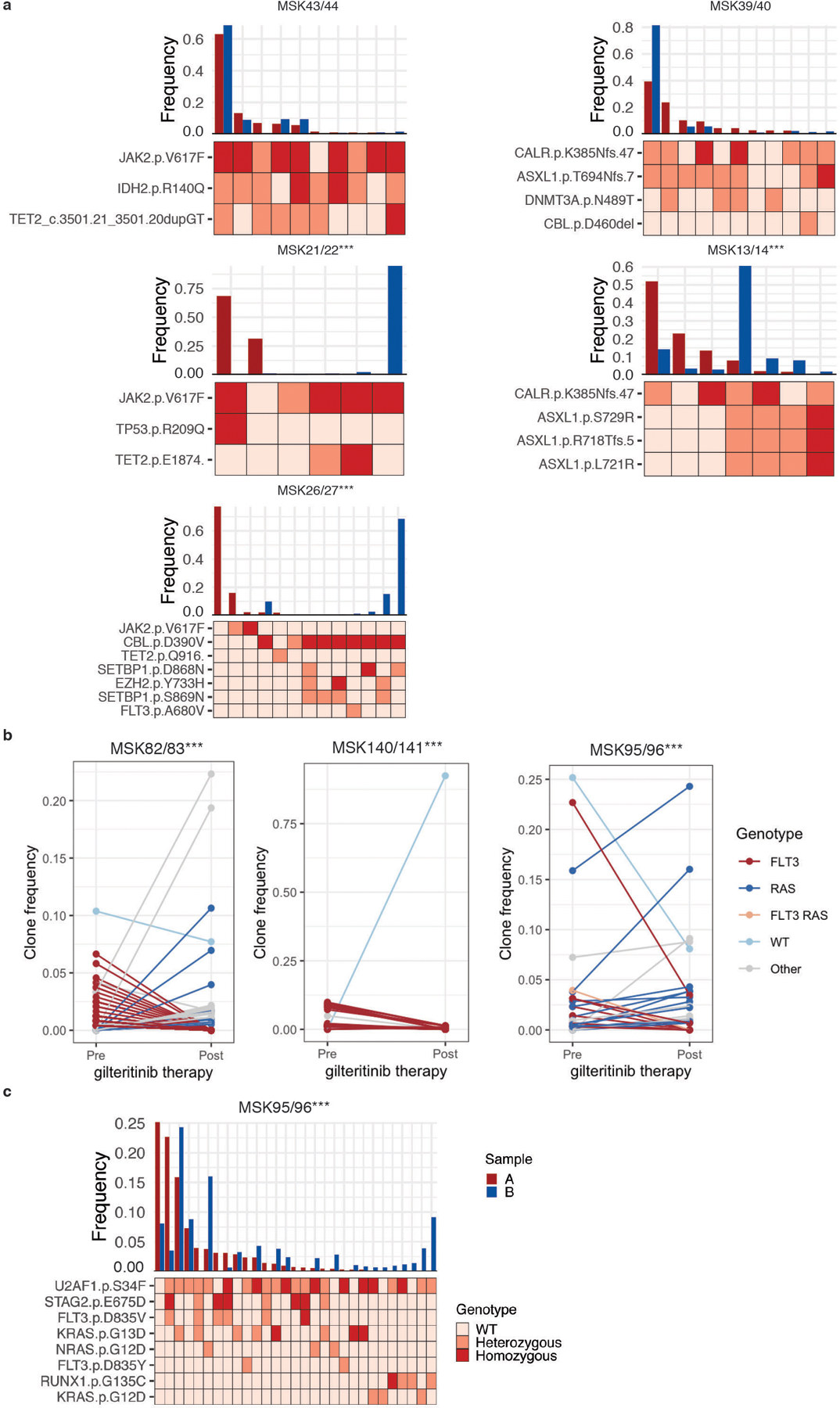
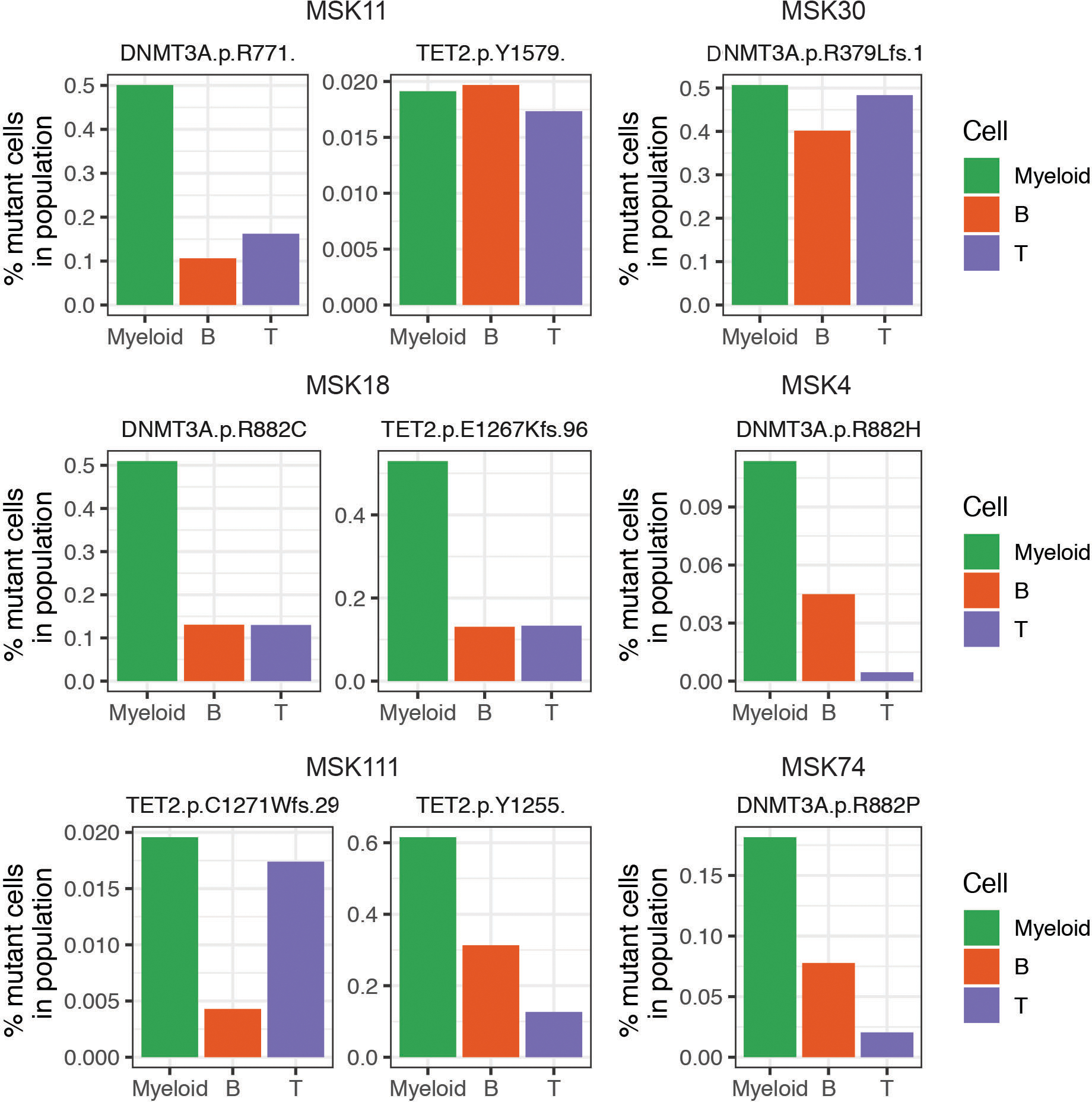
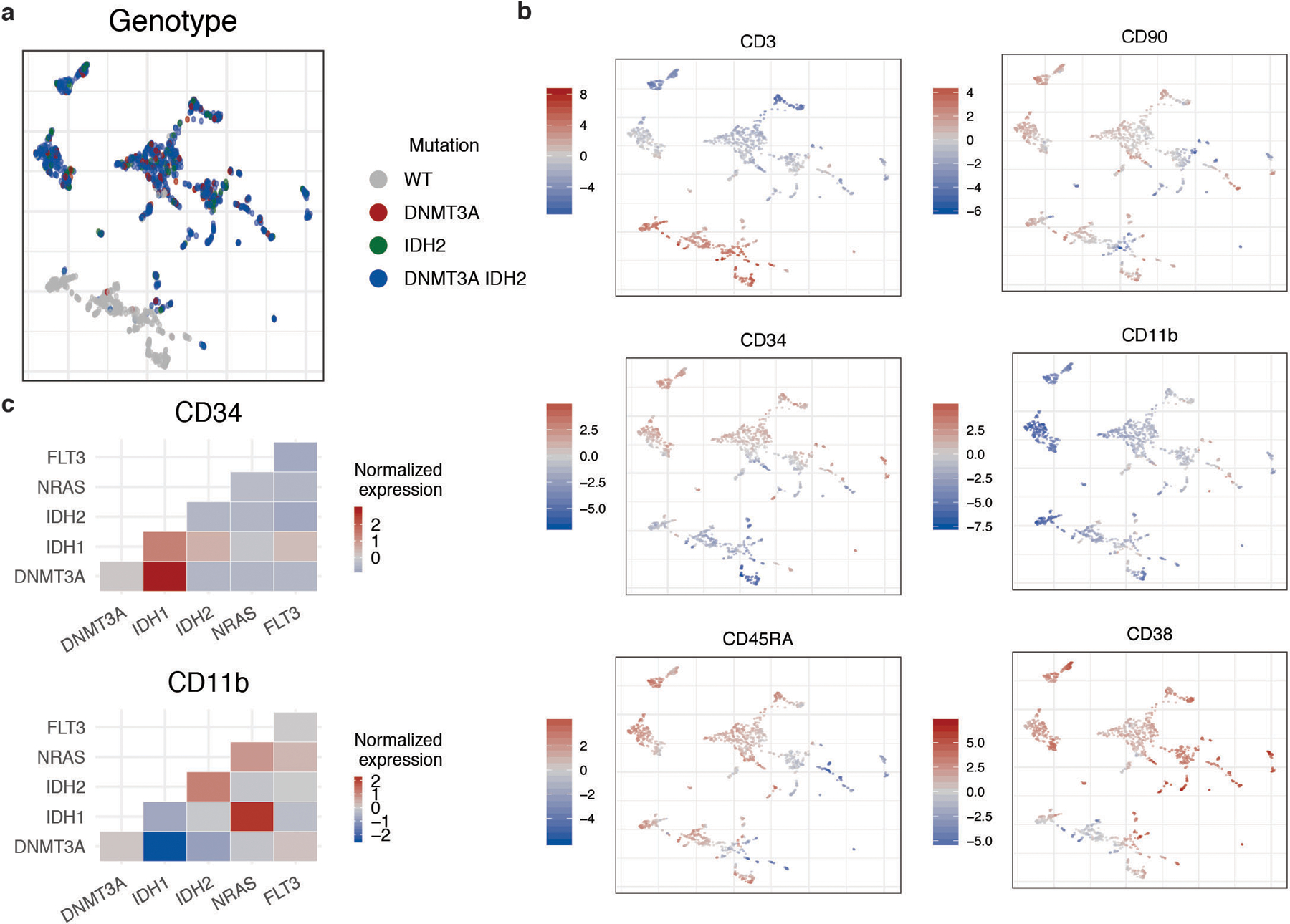
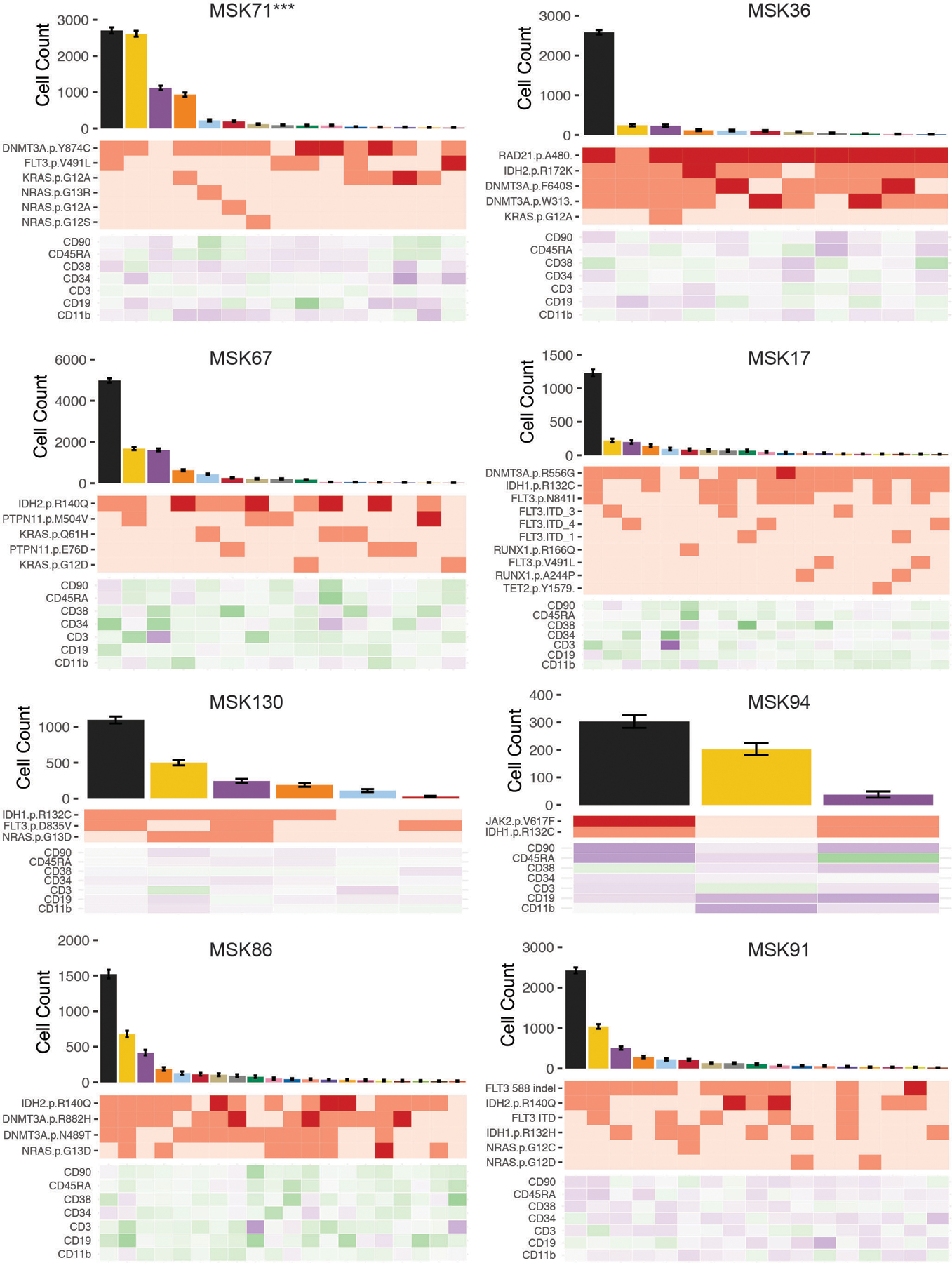
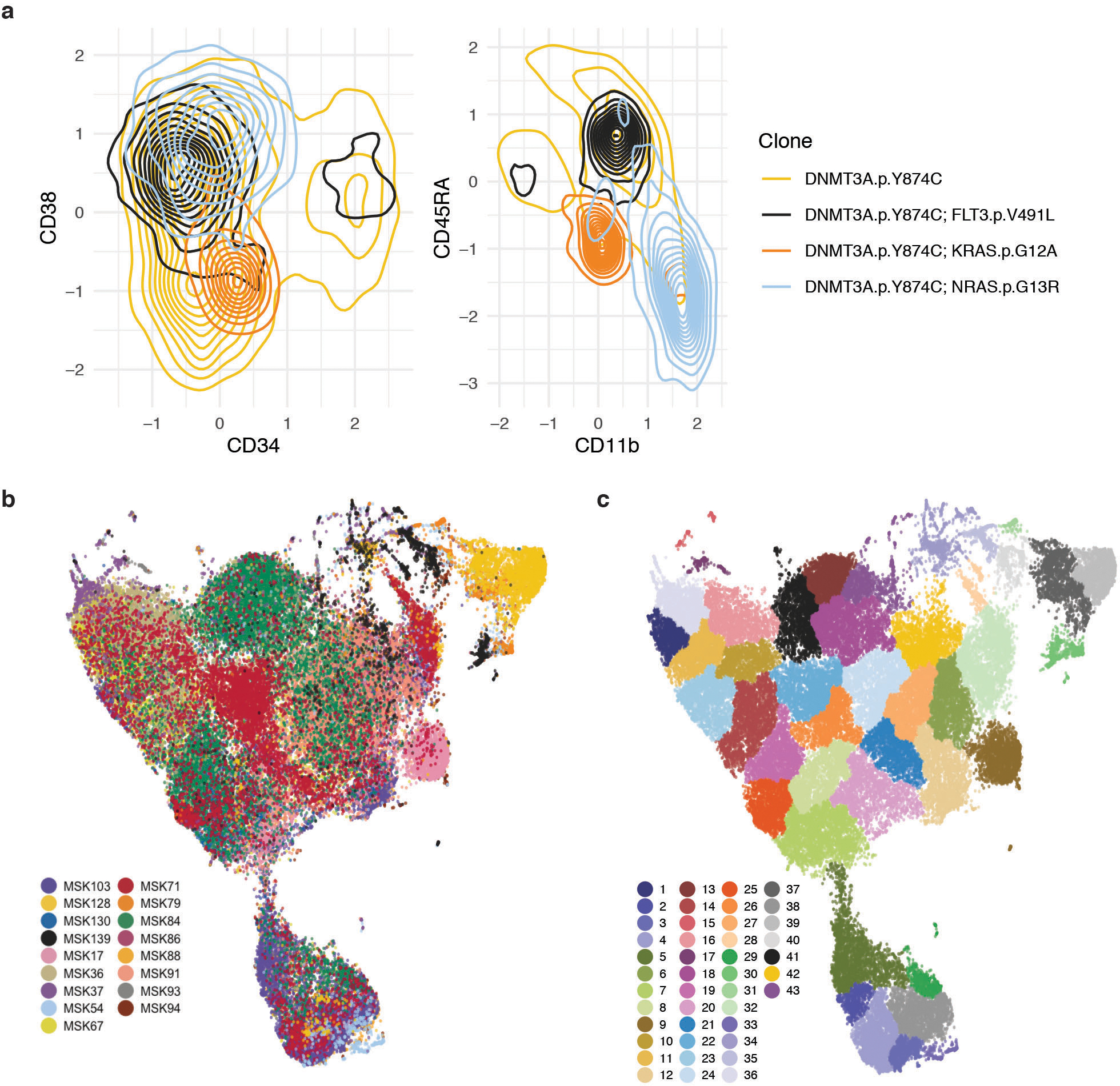
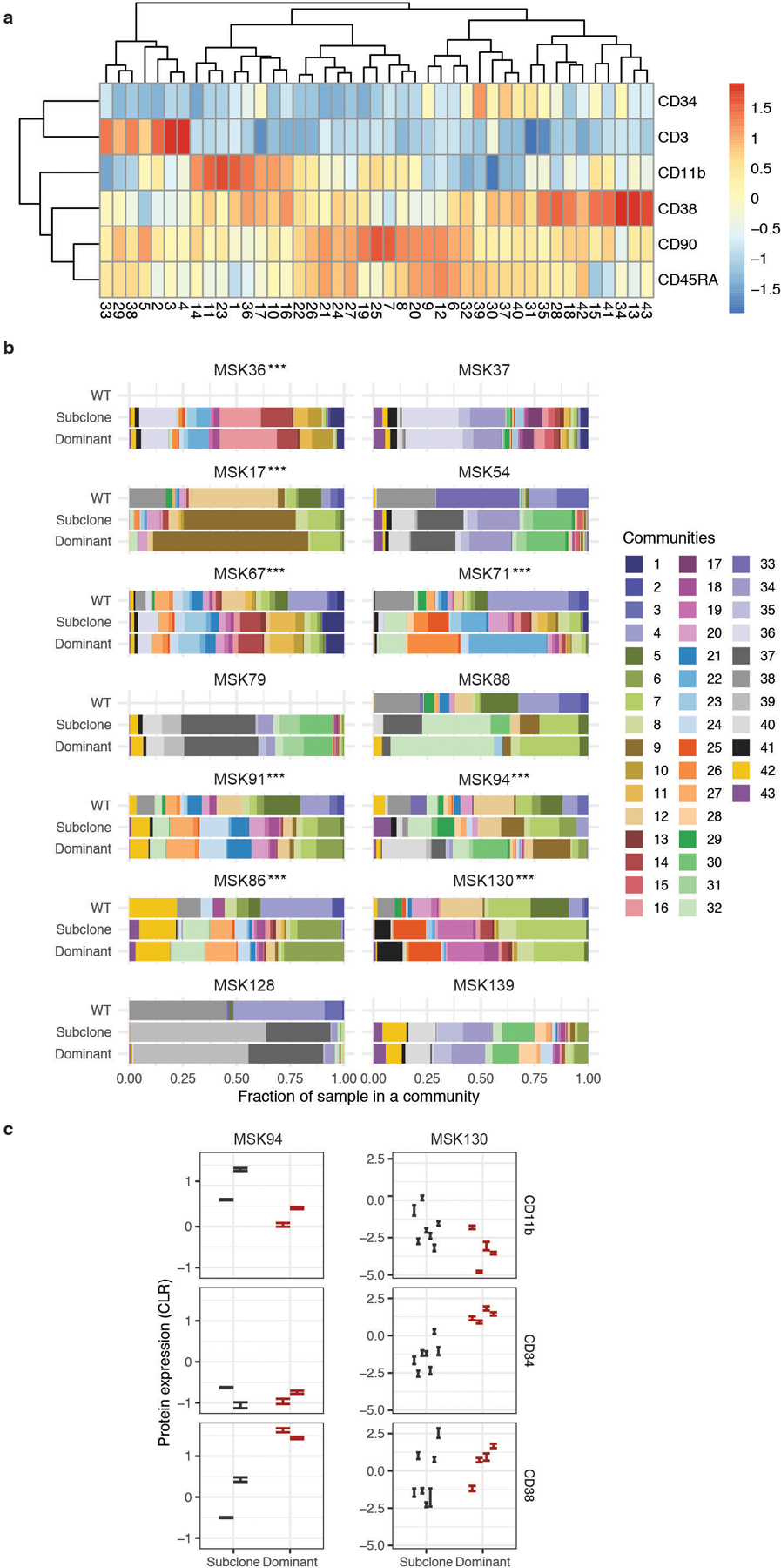
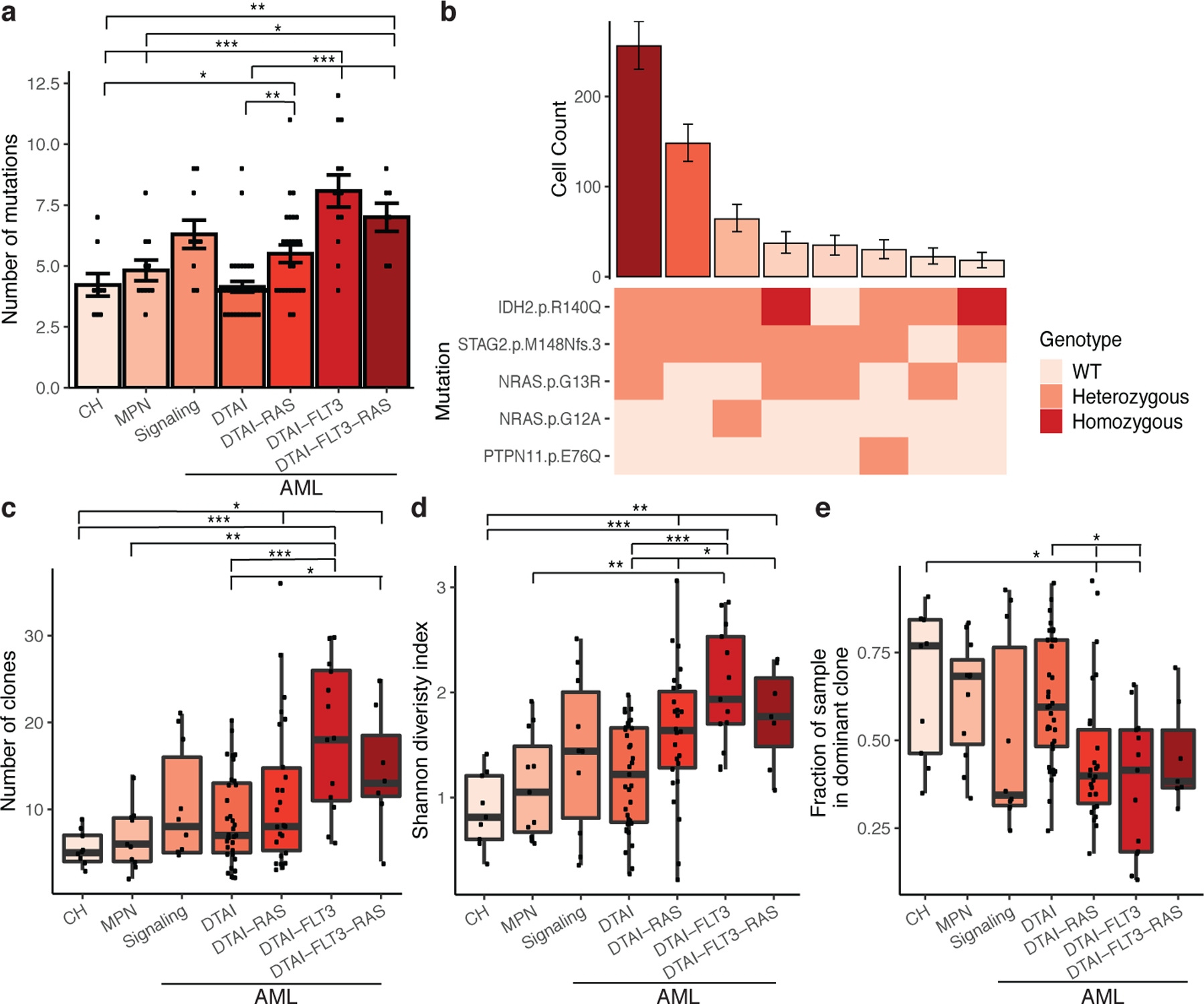
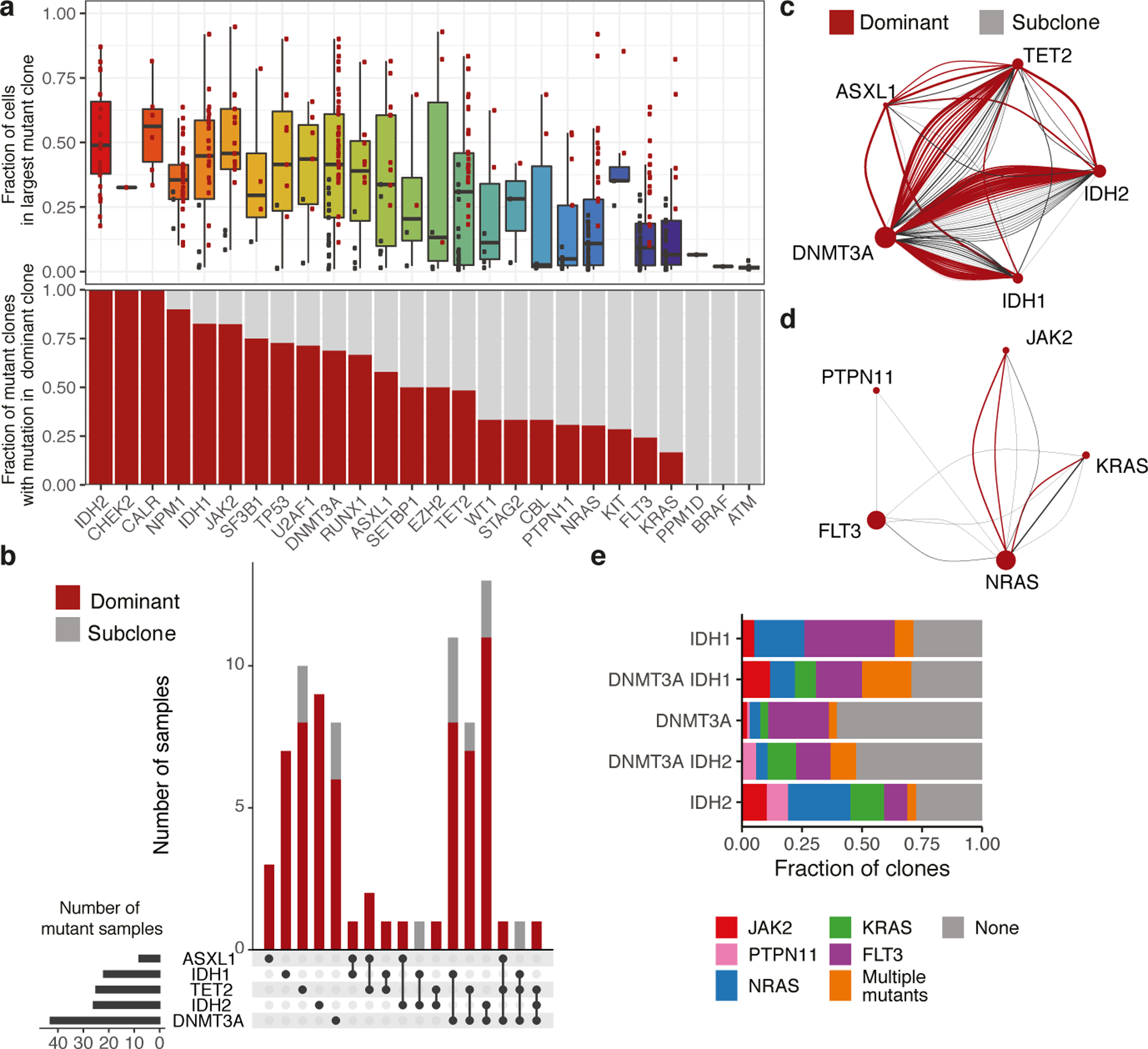
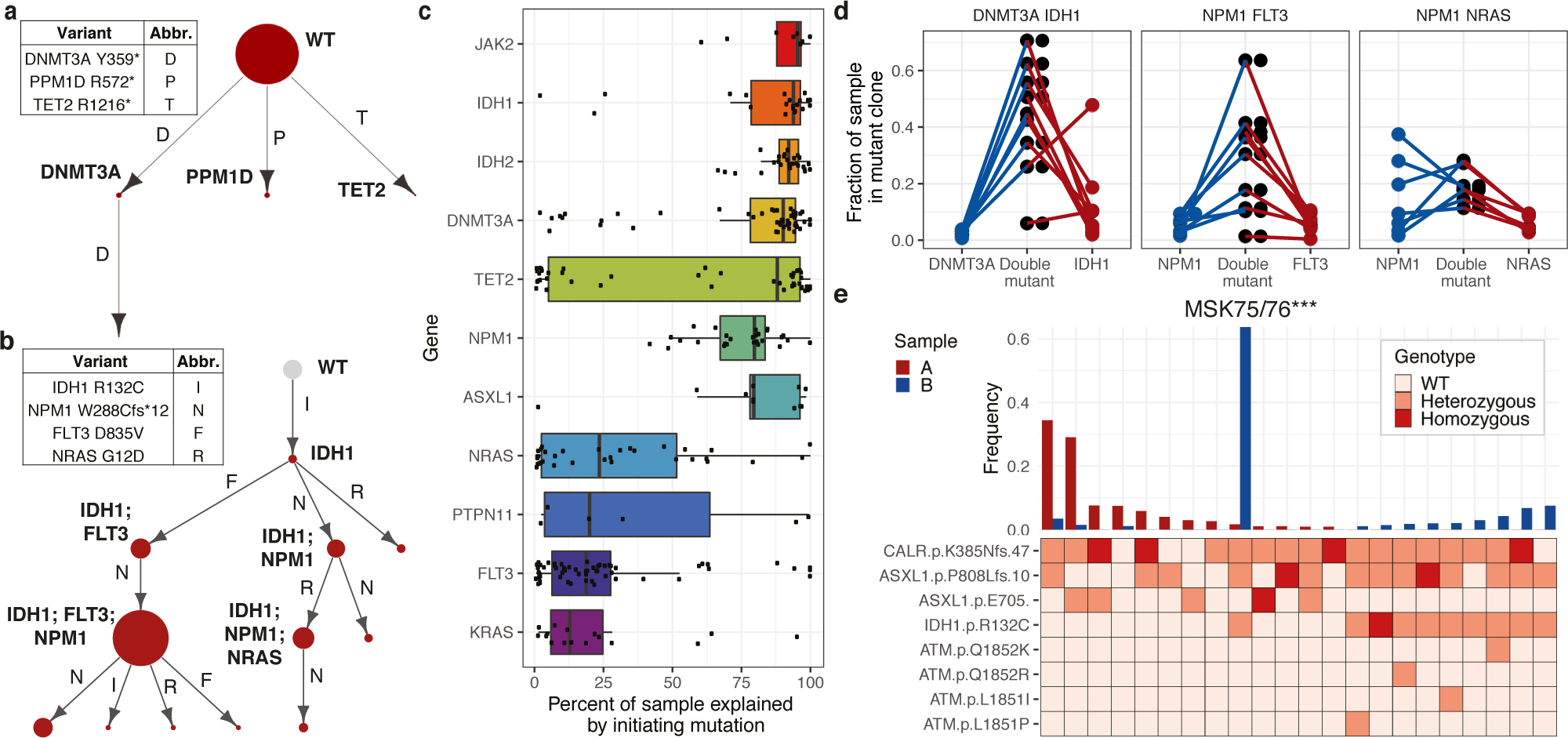
Myeloid malignancies, including acute myeloid leukaemia (AML), arise from the expansion of haematopoietic stem and progenitor cells that acquire somatic mutations. Bulk molecular profiling has suggested that mutations are acquired in a stepwise fashion: mutant genes with high variant allele frequencies appear early in leukaemogenesis, and mutations with lower variant allele frequencies are thought to be acquired later1-3. Although bulk sequencing can provide information about leukaemia biology and prognosis, it cannot distinguish which mutations occur in the same clone(s), accurately measure clonal complexity, or definitively elucidate the order of mutations. To delineate the clonal framework of myeloid malignancies, we performed single-cell mutational profiling on 146 samples from 123 patients. Here we show that AML is dominated by a small number of clones, which frequently harbour co-occurring mutations in epigenetic regulators. Conversely, mutations in signalling genes often occur more than once in distinct subclones, consistent with increasing clonal diversity. We mapped clonal trajectories for each sample and uncovered combinations of mutations that synergized to promote clonal expansion and dominance. Finally, we combined protein expression with mutational analysis to map somatic genotype and clonal architecture with immunophenotype. Our findings provide insights into the pathogenesis of myeloid transformation and how clonal complexity evolves with disease progression.
Conflict of interest statement
Declaration of Interests
L.A.M. and A.D.V. received travel support and honoraria from Mission Bio. A.T.O., R.D.D., P.M., C.A., M.M., and S.S. are employed by Mission Bio and own equity in Mission Bio. A.R.A. is a cofounder and shareholder of Mission Bio. A.Z. has received honoraria from Illumina. M.P.C. has consulted for Janssen Pharmaceuticals. A.D.G. has served on advisory boards or as a consultant for AbbVie, Aptose, Celgene, Daiichi Sankyo, and Genentech, received research funding from AbbVie, ADC Therapeutics, Aprea, Aptose, AROG, Celularity, Daiichi Sankyo, and Pfizer, and received honoraria from Dava Oncology. R.R. has consulted for Constellation, Incyte, Celgene, Promedior, CTI, Jazz Pharmaceuticals, Blueprint, Stemline, Galecto, Pharmessentia, and Abbvie, and received research support from Incyte, Stemline, and Constellation. A.D.V. is on the Editorial Advisory Board of Hematology News. R.L.L. is on the supervisory board of QIAGEN and Mission Bio and is a scientific advisor to Loxo (until Feb 2019), Imago, C4 Therapeutics, and Isoplexis. He receives research support from and consulted for Celgene and Roche and has consulted for Lilly, Jubilant, Janssen, Astellas, Morphosys, and Novartis. He has received honoraria from Roche, Lilly, and Amgen for invited lectures and from Celgene and Gilead for grant reviews. R.L.B., T.R.M., I.S.C., C.F., M.A.P., M.B., B.D., C.L.D., K.B., and S.E.M. disclose no competing interests.
Figures
Comment in
-
Single-Cell Sequencing Reveals Clonal Architecture of Myeloid Cancers.Cancer Discov. 2021 Jan;11(1):13. doi: 10.1158/2159-8290.CD-RW2020-163. Epub 2020 Nov 6. Cancer Discov. 2021. PMID: 33158845
References
Methods References
-
- Cheng DT et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 17, 251–264, doi: 10.1016/j.jmoldx.2014.12.006 (2015). - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
