

Common Adaptive Strategies Underlie Within-Host Evolution of Bacterial Pathogens
- PMID: 33118035
- PMCID: PMC7947768
- DOI: 10.1093/molbev/msaa278
Common Adaptive Strategies Underlie Within-Host Evolution of Bacterial Pathogens
Abstract
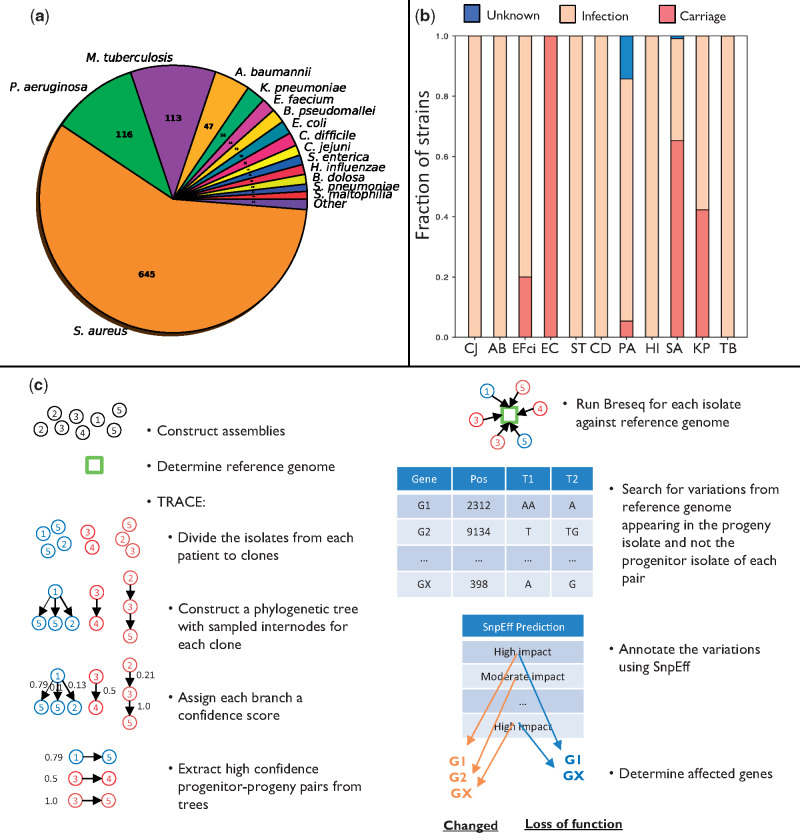
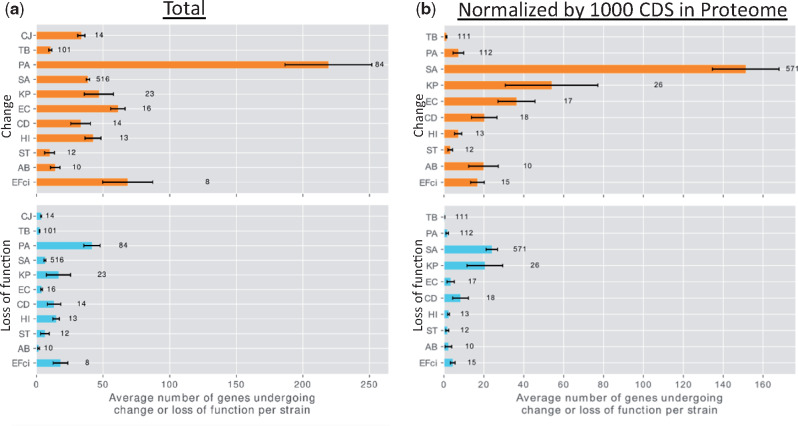
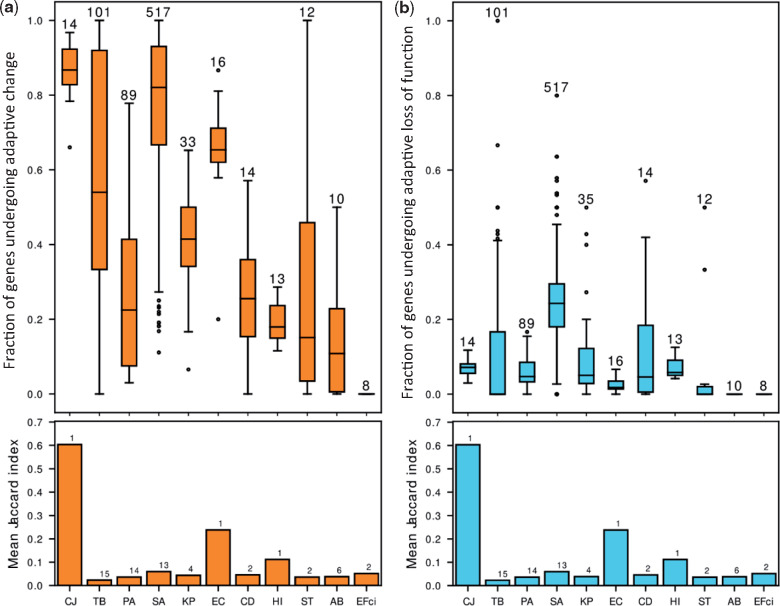
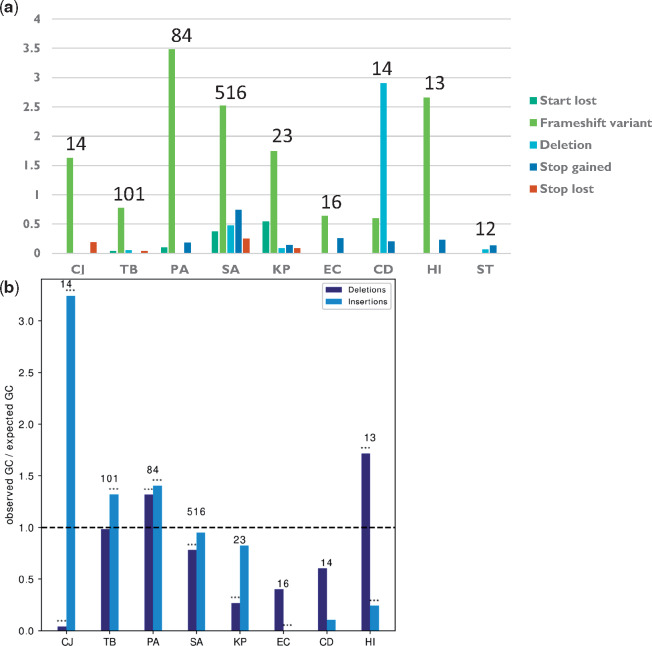
Within-host adaptation is a hallmark of chronic bacterial infections, involving substantial genomic changes. Recent large-scale genomic data from prolonged infections allow the examination of adaptive strategies employed by different pathogens and open the door to investigate whether they converge toward similar strategies. Here, we compiled extensive data of whole-genome sequences of bacterial isolates belonging to miscellaneous species sampled at sequential time points during clinical infections. Analysis of these data revealed that different species share some common adaptive strategies, achieved by mutating various genes. Although the same genes were often mutated in several strains within a species, different genes related to the same pathway, structure, or function were changed in other species utilizing the same adaptive strategy (e.g., mutating flagellar genes). Strategies exploited by various bacterial species were often predicted to be driven by the host immune system, a powerful selective pressure that is not species specific. Remarkably, we find adaptive strategies identified previously within single species to be ubiquitous. Two striking examples are shifts from siderophore-based to heme-based iron scavenging (previously shown for Pseudomonas aeruginosa) and changes in glycerol-phosphate metabolism (previously shown to decrease sensitivity to antibiotics in Mycobacterium tuberculosis). Virulence factors were often adaptively affected in different species, indicating shifts from acute to chronic virulence and virulence attenuation during infection. Our study presents a global view on common within-host adaptive strategies employed by different bacterial species and provides a rich resource for further studying these processes.
Keywords: infectious diseases; microbial genetics; microbiology; within-host adaptation; within-host evolution.
© The Author(s) 2020. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
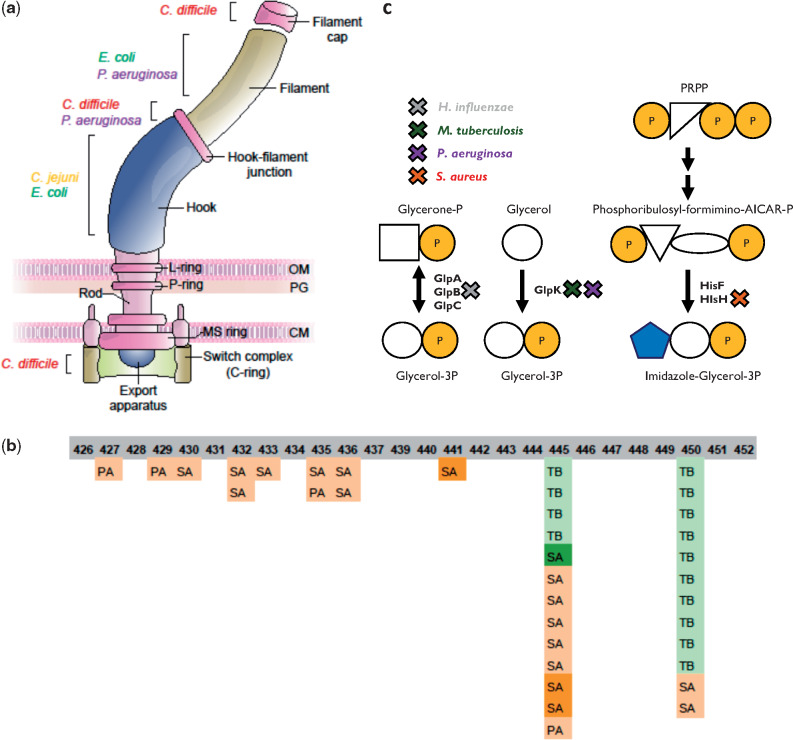
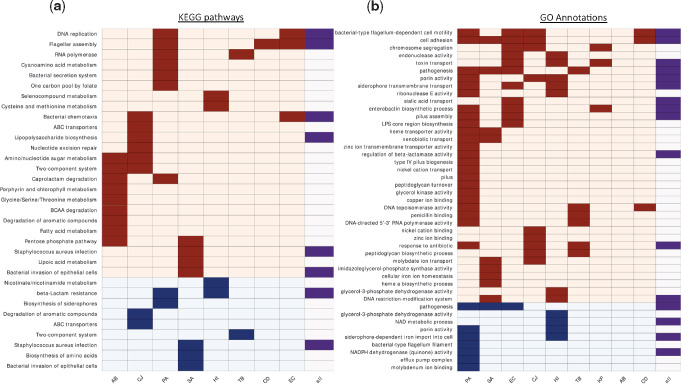
References
-
- Allen RC, Popat R, Diggle SP, Brown SP.. 2014. Targeting virulence: can we make evolution-proof drugs? Nat Rev Microbiol. 12(4):300–308. - PubMed
-
- Andre E, Goeminne L, Cabibbe A, Beckert P, Kabamba Mukadi B, Mathys V, Gagneux S, Niemann S, Van Ingen J, Cambau E.. 2017. Consensus numbering system for the rifampicin resistance-associated rpoB gene mutations in pathogenic mycobacteria. Clin Microbiol Infect. 23(3):167–172. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
